
Abstract
Glycans play an important physiological role and are drawing attention as biomarkers that capture pathophysiological changes. Glycans can be detected by mass spectrometry, but recently matrix-assisted laser desorption/ionization- mass spectrometry imaging (MALDI-MSI) has enabled the visualization of glycans distribution on tissues. In this study, focusing on sialylated glycan (sialoglycans), we investigated the amidation reaction used to visualize glycans distribution, and developed a method of sialic acid derivatization by benzylamidation which is more sensitive than conventional amidation. Furthermore, we adapted this method for visualizing glycans in formalin-fixed paraffin-embedded (FFPE) liver tissue from normal mice and non-alcoholic steatohepatitis (NASH) model mice using MALDI-MSI. As a result, an increase in the distribution of glycan N-Acetylneuraminic acid-(NeuAc) ions was observed in the NASH mouse liver, and the change in glycan structure in the NASH model was clarified.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycans are one of the polysaccharides linked to serine/threonine (O-linked) or asparagine (N-linked) residues in protein [1]. They exist on cell-surface membrane glycoproteins. In many cases, glycans are involved in the regulation of biological processes, indicating that the alteration of glycans might be regarded as valuable biomarkers for various disease states. Thus, glycans can be used as a target molecule for biomarker discovery.
In particular, glycans with sialic acids (sialoglycans) play important roles in biological processes such as cell differentiation, viral infection, and cancer development. Sialic acids often exist on the non-reducing terminal ends of N-glycans or O-glycans. The common sialic acids are N-acetyl neuraminic acid (Neu5Ac) and N-glycolyl neuraminic acid (Neu5Gc). In humans, N-glycolyl neuraminic acids (NeuGc) are not synthesized because the gene encoding cytidine 5′-monophosphate (CMP)-Neu5Ac hydroxide is missing. Some other mammalian species, such as mice, have the gene encoding CMP-Neu5Ac hydroxide, thus NeuGc exists on the glycoprotein.
Glycans can be detected by mass spectrometers, such as electrospray ionization mass spectrometry coupled with high-performance liquid chromatography (LC-ESI MS) and matrix-assisted laser desorption/ionization mass spectrometry (MALDI MS).
Released N-glycans from glycoproteins can be detected by LC-ESI MS, which can elucidate the global glycan structures on the cell surface glycoprotein [2]. On the other hand, recent studies have endorsed the use of MALDI-MS for visualizing the distributions of glycan structures in tissue samples; thus several MALDI-MS imaging (MALDI-MSI) protocols have been developed for visualization in different organs such as the brain, kidney, and liver, and in normal or disease tissues [3,4,5].
Although MALDI-MSI can be used to visualize glycan distribution, the detection of sialoglycans is still a challenge for MALDI-MS. The carboxyl group of the sialic acid often makes positive-mode detection difficult. Negative-mode detection is one of the ways to detect sialoglycans, however, it is not sufficient for sialoglycans. Another way is to neutralize the sialic acids by permethylation [6, 7] esterification [8], hydrazidation [9], or amidation [10, 11] of the carboxyl group. Holst et al. reported an on-tissue sialic acid derivatization method by dimethylamidation and amidation [12]. However, the signal intensities were insufficiently strong to detect sialic acids, so that there might be several undetected glycans.
Here, we developed a method for sialic acids derivatization to visualize glycans (including sialoglycans) on tissue sections with high sensitivity. We report that benzylamidation of the sialic acids is applicable to the released sialo N-glycans from glycoprotein and N-glycans on the tissue surface. The combination of the benzylamidation and MALDI-MSI provided a clear image of sialo N-glycans distribution, enabling us to compare normal mouse liver tissue and non-alcoholic steatohepatitis (NASH) mouse liver tissue [13].
Materials and methods
Chemicals and reagents
Methanol, xylene, ethanol, dimethyl sulfoxide (DMSO), trifluoroacetic acid (TFA) were purchased from FUJIFILM Wako Pure Chemical Industries (Osaka, Japan). Bovine fetuin, 2, 4- dihydroxybenzoic acid (DHB), benzylamine hydrochloride, and aniline hydrochloride were purchased from Sigma-Aldrich (Steinheim, Germany). 1-ethyl-3-(3- (dimethylamino)propyl) carbodiimide (EDC), isopropylamine, and acetohydrazide were purchased from Tokyo Chemical Industry (Tokyo, Japan). (7-Azabenzotriazol-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyAOP) was purchased from Carbosynth Limited (Berkshire, UK). N-glycosidase F (PNGase F) enzyme was purchased from Roche Diagnostics (Mannheim, Germany) or N-zyme Scientifics (Doylestown, PA). Ultrapure water was used for all experiments.
Polyvinylidene difluoride (PVDF) (0.2 μm, for sequencing, 78.4 cm) was purchased from Bio-Rad (Hercules, CA). Cation-exchange columns were made of (30 mg) Dowex 50 W-X8 resin (Supelco, Bellefonte, PA) packed on top of a 10 μl filter tip. The resins were protonated with 1 M HCl (50 μL, 3 times), washed with methanol (50 μL, 3 times), and then equilibrated with water (50 μL, 3 times) before use. Microtiter plates (96-well flat bottom) were purchased from Costar.
Tissue preparation
Three normal mice (C57BL/6 J) and three NASH mice (C57BL/6 J-NASH) (male, 12-week-old) were purchased from Charles River Laboratories Japan Inc. (Kanagawa, Japan). Mouse livers were extirpated and stored in −80 °C until usage. For LC-ESI MS analysis, left lobes of mouse livers were trimmed and snap frozen, then two 100 μm-thick sections were collected and LC-ESI MS analysis was performed. For MALDI-MSI analysis, another left lobe was formalin-fixed and paraffin-embedded (FFPE) and tissue sections were cut at 6 μm thickness, mounted onto ITO-coated glass slides (Bruker Daltonics, Bremen, Germany).
Investigation of the labeling reagent for sialo N-glycan derivatization
Prior to in situ sialic acid derivatization, the nucleophiles for the sialic acid derivatization were investigated. The free N-glycans released from bovine fetuin were used for this investigation. In this study, we selected compounds with benzyl groups for derivatization, expecting high ionization efficiency.
Sialo N-glycan release from bovine fetuin protein
This protocol was modified to reference [2, 14]. Enzymatic release and purification of N-glycans from bovine fetuin was performed as follows:
10 μg of bovine fetuin was dot-blotted onto a polyvinylidene difluoride (PVDF) membrane prewetted with ethanol. After drying at room temperature overnight, the PVDF membrane was washed with ethanol for 1 min and then washed with water three times for 1 min. The protein on the membrane was stained for 5 min with Direct Blue 71 solution (mixture of 800 μl of solution A, 0.1% [w/v] Direct Blue 71 [Sigma-Aldrich]; 10 ml of solution B, acetic acid: ethanol: water = 1:4:5). After destaining with solution B for 1 min, the PVDF membrane was dried at room temperature for 4 h. Proteins stained blue were cut from the PVDF membrane and placed in a 1.5-ml polypropylene tube. The spots were then covered with 500 μl of 1% (w/v) poly (vinylpyrrolidone) 40,000 in 50% (v/v) methanol, agitated for 20 min, and washed 5 times with water (500 μl). PNGase F (2 U in 10 μl of 10 mM NH4HCO3 was added to the tube and incubated at 37 °C for 15 min. An additional 10 μl of water was added to the tube and incubated at 37 °C overnight to release N-glycans. To collect the released N-glycans, the samples were sonicated for 10 min, and the released N-glycans (20 μl) were transferred to new 1.5-ml polypropylene tubes. The sample membrane was washed with water (50 μl twice), and the washings were combined. Combined N-glycans were divided into 20 μL aliquots and used for derivatization as described below.
Hydrolysis of the glycosidic linkages of the sialic acids ilycans
To evaluate the signal intensity of the glycans without sialic acids, the glycosidic bond of the sialic acids was cleaved by acid-hydrolysis treatment as follows:
10 μL of the sialo N-glycans released from fetuin (released sialo N-glycans) was diluted with 10 μL of 10 mM HCl and heated to 80 °C for 2 h. Then 10 μL of 10 mM Tris-HCl buffer (pH 8.0) was added to each sample, and the desialo N-glycans were purified by hydrophilic interaction liquid chromatography (HILIC) using a Sep-Pak NH2 column (Waters, Milford, MA).
Acetohydrazidation
Acetohydrazidation of released sialo N-glycans was performed according to the procedure by Toyoda et al. [9]. A volume of 10 μL of each fetuin glycan solution was added to 50 μL of 1 M acetohydrazide and then adjusted to pH 3 with 1 M HCl. Then, derivatized glycans were purified by HILIC same as above.
Benzylamidation and other amidation
Benzylamidation was performed as follows:
Equivalent amount of 100 pmol of the released sialo N-glycans were added to derivatization solution (125 mM PyAOP, 15% N-MM, and 500 mM of each amine in DMSO). The mixtures were first incubated for 1.5 h at room temperature, then purified by using a Sep-Pak NH2 cartridge, and finally concentrated by freeze-drying. Derivatized glycans were reconstituted in 10 μL of water, and mixed with matrix solution (20 mg/ mL DHB in 50% methanol/0.1% TFA/1 mM NaCl), then they were analyzed by MALDI-TOF MS.
Other amidation reactions were performed using aniline hydrochloride, methylamine-hydrochloride [11], dimethylamine [10], isopropylamine [11] and analyzed same as above.
On-tissue derivatization and N-glycan release for MALDI-MSI
The ITO-coated glass slides were placed onto a heating block at 60 °C for 1 h to enhance adherence of the FFPE sections, sequentially washed in xylene (2 × 10 min), 100% ethanol (2 × 2 min), and 70% ethanol (2 × 2 min) to remove paraffin, and then dried at room temperature for 30 min. Next, on-tissue derivatization was performed by incubating the slides in derivatization solution (125 mM PyAOP, 15% N-MM, 500 mM amine in DMSO) for 1.5 h at room temperature with slow rotation. After derivatization, tissue sections were rinsed with 100% ethanol, sequentially washed with 100% ethanol (2 × 5 min) and water (2 × 5 min) and dried at room temperature (30 min). PNGase F solution (20 μL in 200 μL water) was sprayed onto the ITO-coated slides using an air-brush sprayer (GSI Creos, Tokyo, Japan). N-glycans were released by incubating overnight at 37 °C in a humid environment protected from evaporation. The tissue was covered with matrix solution (20 mg/ mL DHB in 50% methanol/0.1% TFA/1 mM NaCl) using an airbrush sprayer. As another control, the procedure was repeated on subsequent sections, but with other derivatization steps, such as acetohydrazidation, dimethyl amidation, neuraminidase treatment, and without amidation to evaluate the differences between native sialo N-glycans and derivatized sialo N-glycans (or desialo N-glycans) and to assess if the derivatization procedure leads to high detection signal intensities.
MALDI-MS and MS imaging
The MALDI-MS and MALDI-MSI analysis was performed on a UltrafleXtreme MALDI-TOF/TOF instrument (Bruker Daltonics) in positive ion reflectron mode. The conditions were set as follows: m/z range, 700 to 4000. For imaging, the following conditions were set: 1000 laser shots per pixel, and a 100 μm × 100 μm spatial resolution. MALDI data acquisition was enabled by using the Flex software suite (flexControl, flexImaging 4.0, flexAnalysis 3.4; Bruker Daltonics).
LC-ESI MS for analysis of glycan alditols
Frozen tissue sections cut by Cryostat CM1950 (Leica Biosystems) were homogenized by pipetting in lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, containing protease inhibitor, pH 7.4). The homogenized tissues were centrifuged (760 g, 4 °C, 20 min), then the supernatants were ultracentrifuged (120,000 g, 4 °C, 80 min). The pellets were resuspended in TritonX-114 solution (50 mM Tris-HCl, 100 mM NaCl, containing 1% TritonX-114, pH 7.4) by vigorous pipetting, and the resuspensions were incubated first for 10 min on ice then for 20 min at 37 °C and finally centrifuged (1940 g, 2 min). The detergent layers were transferred to new 1.5 mL tubes, then after adding cold acetone, incubated −30 °C for overnight and finally spun down (1940 g, 2 min). The resulting pellets were used for this study as membrane proteins.
The precipitated membrane proteins were dissolved with 10 μL of 8 M urea. The solubilized proteins were dot-blotted (4 times with 2.5 μL) onto a PVDF membrane prewetted with ethanol. After drying at room temperature overnight, the PVDF membrane was washed with ethanol for 1 min, washed with water three times for 1 min, stained for 5 min with Direct Blue 71 for protein visualization (800 μl solution A: 0.1% [w/v] Direct Blue 71 [Sigma-Aldrich] in 10 ml solution B: acetic acid: ethanol: water = 1:4:5). After destaining with solution B for 1 min, the PVDF membrane was dried at room temperature for 4 h. N-glycans were released from the dot-blotted proteins and then were reduced to N-glycan alditols essentially by using the method of Nakano et al. [2].
Dried N-glycan alditols were resuspended in 10 mM NH4HCO3 (15 μL) immediately before LC-ESI MS analyses. N-glycan alditols were separated using a porous graphitized carbon column (5 μm HyperCarb, 100 × 1.0 mm ID, Thermo Fisher Scientific). Separation of N-glycan alditols was achieved using a sequence of isocratic and two segmented linear gradients: 0–8 min, 10 mM NH4HCO3; 8–38 min, 6.75–15.75% (v/v) CH3CN in 10 mM NH4HCO3; 38–73 min, 15.75–40.5% (v/v) CH3CN in 10 mM NH4HCO3 (flow rate: 50 μL/min). With regard to the mass spectrometer (LTQ Orbitrap XL, a hybrid linear ion trap-orbitrap mass spectrometer; Thermo Fisher Scientific), the voltage of the capillary source was set at 3 kV, and the temperature of the transfer capillary was maintained at 300 °C. The capillary voltage and tube lens voltage were set at −18 V and − 110 V, respectively. MS spectra were obtained using the orbitrap detector in negative ion mode (mass range, m/z 500 to m/z 2500; resolution, 15,000; mass accuracy, 5 ppm), and MS/MS spectra were obtained using the ion trap detector (data dependent top 3, CID). Monoisotopic masses of glycans observed in MS were computed to find possible monosaccharide compositions using the GlycoMod tool available on the ExPASy server (http://au.expasy.org/tools/glycomod; mass tolerance for precursor ions, ± 0.01 Da). Xcalibur software ver. 2.2 (Thermo Fisher Scientific) was used to display the base peak chromatogram (BPC) and extracted ion chromatogram (EIC) and to analyze MS and MS/MS data.
Results and discussion
Investigation of the derivatization method using sialoglycans released from bovine fetuin
We aimed to develop a new, more sensitive derivatization method than the conventional method, that could be used for MALDI-MS imaging of sialo N-glycans. We investigated the labeling reagents which can ionize sialo N-glycans efficiently. Prior to on-tissue sialylation, we tested new labeling reagents (benzylamine and aniline) for N-glycan sialylation, and we also tested the conventional methods to compare the signal intensities of the products of MALDI ionization. Released sialo N-glycans from bovine fetuin were used because their structures have been identified and all of them had a terminal Neu5Ac [15, 16].
For carboxylic acid activator, we used (7-azabenzotriazol-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyAOP) because PyAOP labels the sialic acid amines efficiently, forming α2, 3- and α2, 6-linkages in one step. The glycans were labeled with amines or acetohydrazide and detected by MALDI-MS in positive ion mode.
As a result, each amidation reaction enabled us to detect the sialo N-glycans of fetuin (mono or bi-sialylated bi-antennary glycans, and mono, bi, tri, or tetra-sialylated tri-antennary glycans, a total of 6 peaks including the isomers), however, the native-form of sialo N- glycans was not detected. The benzylamide forms and phenylamide forms of sialo N-glycans were detected with a theoretical mass shift (benzylamide: +89.1 Da, phenylamide: +75.1 Da), indicating that sialoglycans were successfully amidated with PyAOP reagent, and the derivatized sialo N-glycans could be detected by MALDI- MS.
Table 1 shows the signal intensities of tri-sialylated tri-antennary glycans. The signal intensities of the benzylamide form and phenylamide form were compared to those of forms obtained by conventional methods (with acetohydrazide, methylamine, dimethylamine, and isopropylamine as labeling reagents) and also the hydrolyzed form (i.e., sialic acids removed by acid hydrolysis). The signal intensities of the acetohydrazide, methylamide, dimethylamide, or isopropylamide forms of sialylated N-glycans were quite low. On the other hand, intensity was 5.6-fold higher for the benzylamide form than the acetohydrazide form. Compared to the hydrolyzed form (See Table 2), the benzylamide form showed almost the same intensity (Relative intensity: 1.1), indicating that the benzylamide form had not been affected by ionization suppression of the sialic acids. Thus, MALDI-MS can detect the benzylamide form with higher sensitivity than conventional forms. The signal intensity of the phenylamide form was also higher than that of conventional forms. Aromatic compounds are generally stable thus their molecular ions usually show high signal intensity. Linking aromatic compounds to sialoglycans raises the signal intensities of their cations, compared to the other labeling methods. However, the intensity of the phenylamide form was much lower than that of the benzylamide form of sialo N-glycans. Therefore, we adopted benzylamide as our labeling reagent for the following MALDI-MSI experiment (reaction scheme, see Fig. 1, Sialic acid-Galactose- Derivatized sialic acid-Galactose-).

Reaction scheme of sialic acid benzyl amidation
To confirm and visualize the distribution of sialo N-glycans on the tissue surface, we next performed MALDI-Mass Spectrometry Imaging (MALDI-MSI) using normal and NASH mouse livers and performed MALDI-MS to identify the method of labeling producing the most detectable signal.
Profiling of N-glycan in the mouse liver tissue by LC-ESI MS
To identify and profile N-glycans in mouse livers, we performed LC-ESI MS measurement by using the LTQ Orbitrap XL (ThermoFisher Scientific). N-glycans on the normal/NASH liver cell membrane proteins were extracted from two liver cryosections (approximately 60 mg of tissue) and detected by LC-ESI MS analysis as N-glycan alditols.
The base peak chromatograms (BPC) of the separated N-glycans showed a difference in global distribution between the normal liver and NASH liver. Each N-glycan was associated with its own peak (Fig. 2) and a total of 51 N-glycans were identified (summarized in Table 3 LC-ESI MS).

N-Glycan identification by LC- ESI MS. The base peak chromatogram of N-glycans in normal mouse liver (above) and NASH mouse liver (below)
These glycan structures included the high-mannose type, asialo-, monosialo-, disialo-, trisialo-, and tetrasialo-N-glycans with or without fucosylation. High-mannose type glycans and disialo-bi-antennary N-glycan, which contains Neu5Gc at each non-reducing terminal, were observed in large amounts in each liver. To analyze the difference between normal and NASH livers, we compared the extracted ion chromatogram (EIC) of N-glycans that seemed to be altered in the NASH sample (Fig. 3). Compared to normal liver, NASH liver showed increased N-glycans containing NeuAc or fucose and decreased N-glycans containing NeuGc.

Signal comparison of the N-glycans identified by LC-ESI MS. The base peak (a) or extracted ion (b-j) chromatograms of N-glycans in NASH mouse liver (above) and normal mouse liver (below)
MALDI-MSI detection of in situ benzylamidated sialoglycans
In order to demonstrate coarse MS measurement of the on-tissue N-glycans, FFPE normal mouse liver sections were used for MALDI-MSI data acquisition. The on-tissue glycan derivatizations have been successfully performed by incubating the slides with tissue sections immersed in derivatization solution. Compared to the non-enzymatic treatment, the PNGase F treatment resulted in several peaks assigned putative identities as N-glycans (putative glycan peaks). All putative glycan peaks were detected as sodium adducts. Figure 4 shows the peaks detected after acetohydrazidation, dimethylamidation, or benzylamidation. From LC-ESI MS measurement described above, 2 NeuGc-biantennary glycans (Fig. 3, 13a) detected with the highest intensity as sialoglycans, thus we compared the intensities of derivatized 2 NeuGc-biantennary glycans. As a result, benzylamidation derivatives analysis clearly showed peaks that seemed to be good S/N ratio. The intensities and numbers of detected peaks were clearly larger when derivatization is by benzylamidation than by other methods (Fig. 4C), especially peaks with m/z values >2000.

Comparison of the labeling reagent for MALDI-MSI detection of N-glycans on the normal mouse liver. Sialoglycans on FFPE normal mouse liver tissue were labeled with (a) acetohydrazide, (b) dimethylamine, or (c) benzylamine, and the signal intensities of the 2 NeuGc-biantennary glycans were compared on the same scale
Comparison of detected N-glycan between normal and NASH mouse livers
Table 3 provides a combined list of the N-glycans which were identified and characterized by LC-ESI MS profiling and further analyzed by on tissue-derivatization MALDI-MSI. The proposed compositions and structures are provided as well as the calculated [M-2H]2− from GlycoMod (web tool). These results showed there were overlaps of observed N-glycans for both measurement methods. The majority of the N-glycans were sialylated forms, thus the sialic acid derivatization was effective for liver tissues. In MALDI-MSI, the benzylamidation led to the detection of 13 distinct sialo N-glycans, which was more than acetohydrazidation (only 1 sialo N-glycan detected), indicating that the benzylamidation can be a powerful method of on-tissue N-glycan sialylation. Including asialo N-glycans, a total of 21 N-glycans were detected (summarized in Table 3 MALDI-MSI), indicating that asialo N-glycans are also detectable through derivatization.
Next, comparing the mass spectrum of sialo N-glycans between normal and NASH mouse livers, peaks at m/z 2423.5, m/z 2439.5, m/z 2455.5, m/z 2221.5, m/z 2570.0, m/z 2586.0, m/z 2602.0, and m/z 2367.5 were shown to be altered in NASH liver. The sialo N-glycans were visualized using Fleximaging software (Fig. 5).

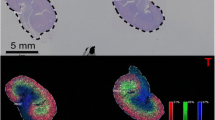
MALDI-MSI of the N-glycans on the normal and NASH mouse FFPE livers. (a) Optical image of the tissue slices (above: left median lobe of the NASH mouse, below: left median lobe of the normal mouse) on an ITO-coated glass slide. Area surrounded by white dotted line was measured by MALDI-MSI measurement. (b) 2 NeuAc-bi antennary; (c) 1 NeuAc and 1 NeuGc-biantennary; (d) 2 NeuGc-biantennary; (e) 1 NeuGc-triantennary; (f) high-mannose; (g), (h), (i), and (j) are (b), (c), (d), and (e), respectively, with fucosylation
2 NeuGc-biantennary glycans with ions at m/z 2455.5 were the most abundant of all glycans in both livers and their abundance was greater in normal liver. On the other hand, 1 NeuAc and 1 NeuGc-biantennary glycan, and 2 NeuAc-biantennary glycans (m/z 2439.5 and m/z 2423.5) were relatively increased in NASH liver compared to normal liver. These results indicate that the abundance of Neu5Ac was increased in NASH liver.
The fucosylated sialo N-glycans (m/z 2570.0, m/z 2586.0, m/z 2602.0, and m/z 2367.5) were also increased in NASH liver, compared to the glycans without fucosylation (m/z 2423.5, 2439.5, 2455.5, and 2221.5). There are some reports that N-glycan fucosylation is related to cancer [17], and that fucosylation seemed to be important in NASH progression to cancer.
Conclusion
In this study, we reported a new derivatization method which enables us to detect on-tissue sialo N-glycans with high sensitivity. Benzylamide forms of sialo N-glycans were successfully detected by MALDI-MSI with benzylamidation affording higher signal intensity than previous derivatization methods. Using this method, we could visualize several sialo N-glycans and alteration of their levels in normal and NASH mouse livers. We expect our method will be used to obtain important biological information such as change in glycobiomarker distribution during disease development, which can be monitored by this technique.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- MALDI -MSI:
-
matrix-assisted laser desorption/ionization- mass spectrometry imaging
- FFPE:
-
formalin-fixed paraffin-embedded
- NASH:
-
non-alcoholic steatohepatitis
- NeuAc:
-
N-Acetylneuraminic acid
- NeuGc:
-
N-glycolylneuraminic acids
- DHB:
-
dihydroxybenzoic acid
- EDC:
-
1-ethyl-3-(3- (dimethylamino)propyl) carbodiimide
- PyAOP:
-
(7-Azabenzotriazol-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate
References
Varki A, Cummings RD, Esko JD, et al., Essentials of Glycobiology 3rd edition. Cold Spring Harbor (NY): 2015-2017
Nakano, M., Saldanha, R., Göbel, A., Kavallaris, M., Packer, N.H.: Identification of glycan structure alterations on cell membrane proteins in desoxyepothilone B resistant leukemia cells. Mol. Cell. Proteomics. 10, M111.009001 (2011)
Powers, T.W., Jones, E.E., Betesh, L.R., Romano, P.R., Gao, P., Copland, J.A., Mehta, A.S., Drake, R.R.: Matrix assisted laser desorption ionization imaging mass spectrometry workflow for spatial profiling analysis of N-linked glycan expression in tissues. Anal. Chem. 85, 9799–9806 (2013)
Powers, T.W., Neely, B.A., Shao, Y., Tang, H., Troyer, D.A., Mehta, A.S., Haab, B.B., Drake, R.R.: MALDI imaging mass spectrometry profiling of N-glycans in formalin-fixed paraffin embedded clinical tissue blocks and tissue microarrays. PLoS One. 9, e106255 (2014)
Gustafsson, O.J., Briggs, M.T., Condina, M.R., Winderbaum, L.J., Pelzing, M., McColl, S.R., Everest-Dass, A.V., Packer, N.H., Hoffmann, P.: MALDI imaging mass spectrometry of N-linked glycans on formalin-fixed paraffin-embedded murine kidney. Anal. Bioanal. Chem. 407, 2127–2139 (2015)
Kang, P., Mechref, Y., Klouckova, I., Novotny, M.V.: Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun. Mass Spectrom. 19, 3421–3428 (2005)
Zaia, J.: Mass spectrometry and Glycomics. OMICS. 14, 401–418 (2010)
Reiding, K.R., Lonardi, E., Hipgrave Ederveen, A.L., Wuhrer, M.: Ethyl esterification for MALDI-MS analysis of protein glycosylation. Methods Mol. Biol. 394, 151–162 (2016)
Toyoda, M., Ito, H., Matsuno, Y.K., Narimatsu, H., Kameyama, A.: Quantitative derivatization of sialic acids for the detection of sialoglycans by MALDI MS. Anal. Chem. 80, 5211–5218 (2008)
de Haan, N., Reiding, K.R., Haberger, M., Reusch, D., Falck, D., Wuhrer, M.: Linkage-specific sialic acid derivatization for MALDI-TOF-MS profiling of IgG glycopeptides. Anal. Chem. 87, 8284–8291 (2015)
Nishikaze, T., Tsumoto, H., Sekiya, S., Iwamoto, S., Miura, Y., Tanaka, K.: Differentiation of Sialyl linkage isomers by one-pot Sialic acid Derivatization for mass spectrometry-based glycan profiling. Anal. Chem. 89, 2353–2360 (2017)
Holst, S., Heijs, B., de Haan, N., van Zeijl, R.J., Briaire-de Bruijn, I.H., van Pelt, G.W., Mehta, A.S., Angel, P.M., Mesker, W.E., Tollenaar, R.A., Drake, R.R., Bovée, J.V., McDonnell, L.A., Wuhrer, M.: Linkage-specific in situ Sialic acid Derivatization for N-glycan mass spectrometry imaging of formalin-fixed paraffin-embedded tissues. Anal. Chem. 88, 5904–5913 (2016)
Matsumoto, M., Hada, N., Sakamaki, Y., Uno, A., Shiga, T., Tanaka, C., Ito, T., Katsume, A., Sudoh, M.: An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 94, 93–103 (2013)
Wilson, N.L., Schulz, B.L., Karlsson, N.G., Packer, N.H.: Sequential analysis of N- and O-linked glycosylation of 2D-PAGE separated glycoproteins. J. Proteome Res. 1, 521–529 (2002)
Green, E.D., Adelt, G., Baenziger, J.U., Wilson, S., Van Halbeek, H.: The asparagine-linked oligosaccharides on bovine fetuin. Structural analysis of N-glycanase-released oligosaccharides by 500-megahertz 1H NMR spectroscopy. J. Biol. Chem. 263, 18253–18268 (1988)
Townsend, R.R., Hardy, M.R., Hindsgaul, O., Lee, Y.C.: High-performance anion-exchange chromatography of oligosaccharides using pellicular resins and pulsed amperometric detection. Anal. Biochem. 174, 459–470 (1988)
Nakano, M., Nakagawa, T., Ito, T., Kitada, T., Hijioka, T., Kasahara, A., Tajiri, M., Wada, Y., Taniguchi, N., Miyoshi, E.: Site-specific analysis of N-glycans on haptoglobin in sera of patients with pancreatic cancer: a novel approach for the development of tumor markers. Int. J. Cancer. 122, 2301–2309 (2008)
Author information
Authors and Affiliations
Contributions
M.N. and K.M. conceived and designed the experiments; T.S. and A.W. performed the experiments; T.S., A.W., M.N., and K.M. analyzed the data; T.S., M.N., and K.M. wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Saito, T., Watanabe, A., Nakano, M. et al. MALDI-TOF mass spectrometry imaging for N-glycans on FFPE tissue sections of mouse NASH liver through Sialic acid Benzylamidation. Glycoconj J 38, 167–175 (2021). https://doi.org/10.1007/s10719-021-09984-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-021-09984-w