
The effect of mechanochemical activation on the formation of a cobalt ferrite phase from iron and cobalt oxalates was studied. X-ray phase, x-ray diffraction, and simultaneous thermal analysis, scanning electron microscopy, and the low-temperature nitrogen adsorption-desorption method were used to study the composition and properties of the resulting product. It was established that on solid-phase interaction of the initial components cobalt ferrite is formed at 1100°C. Preliminary mechanochemical activation of the initial components in a roll-ring vibratory mill makes it possible to reduce the temperature of cobalt ferrite synthesis to 400°C. The properties of cobalt ferrite can be improved by lowering its synthesis temperature. It is shown that CoFe2O4 obtained by the mechanochemical method of synthesis has a more developed specific surface area and porous structure compared to cobalt ferrite obtained by solid-phase interaction of the initial components without pretreatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Cobalt ferrite draws the attention of many researchers, as it has a set of unique properties allowing it to be used in different industries. Cobalt ferrite nanoparticles are widely used as magnetic materials [1, 2], biosensors [1], ferromagnetic liquids [1, 2], catalysts, and photocatalysts [1, 2].
It is well known that materials used in different industries must have different properties. Ferrites used as catalysts should have a large surface area and a developed porous structure as well as the necessary set of structural and mechanical properties. The method by which it is obtained makes a large contribution to the properties of the final product. Different methods are used to synthesize cobalt ferrite: solid-phase synthesis [1, 2], coprecipitation [1], hydrothermal [1, 2], and sol-gel [3, 4] as well as the Massart method [1] and microwave synthesis [1, 2]. These methods have a number of disadvantages, for example, solid-phase synthesis requires high temperatures and prolonged calcination, so that the product cannot be obtained in a highly dispersed state. Methods based on the production of ferrites from salt solutions are very laborious, multistage, and require strict control of process parameters (temperature, pH, and others. In addition, in the course of their implementation a large amount of wastewater is generated, which requires disposal. These parameters make a large contribution to the properties and cost of the final product. Most of these shortcomings can be eliminated by using mechanochemical synthesis. The main advantages of this method are ease of implementation and the possibility of obtaining a large amount of nanostructured powders in a short period of time [1, 2].
Quite many works [3,4,5,6, – 7] are devoted to the issues of mechanochemical synthesis of ferrites; intense research has persisted in recent years. The authors of [8] propose to synthesize cobalt ferrite nanoparticles in two stages: coprecipitation and mechanochemical comminution of coprecipitation precursors. A similar method was used in [9] to obtain Cu–Co ferrites. In [10] it is proposed to obtain cobalt ferrite, deposited on a SiO2 base, by joint mechanical activation and subsequent heat treatment of the system 6Fe2O3–2Co3O4–Si. In [11] it is proposed to obtain CoFe2O4 by mechanical grinding of pre-prepared layered double cobalt hydroxocarbonate CoFe2(OH)4(CO3)2∙ nH2O. The use of cobalt hydroxide Co(OH)2 and iron oxalate FeC2O42∙ H2O as precursors for the mechanochemical synthesis of cobalt ferrite is proposed in [11].
Analyzing the work on producing highly dispersed cobalt ferrites under nonequilibrium conditions, it can be concluded that the use of mechanochemical synthesis has a positive effect on the conditions for the formation of crystalline cobalt ferrite as well as its properties.
As noted earlier, ferrites are widely used in catalysis. They are catalysts for a number of processes, such as catalytic oxidation of chlorobenzene [6], decomposition of cyclohexane [1], dehydration of ethylbenzene [1], oxidation of alkenes [2], and others. A promising application for them is to neutralize gaseous emissions, since ferrites can successfully replace catalysts containing precious metals such as platinum, palladium, gold, and others [3].
An analysis of the published data shows [12] that cobalt ferrite is a promising material that can find wide applications, but the fundamental data on the effect of precursors on the formation of the crystalline phase of cobalt ferrite and its physicochemical and catalytic properties are sparse in the literature. For this reason, the objectives of the present work are to investigate interaction in the system CoC2O42 ∙ H2O – FeC2O42 ∙ H2O on joint mechanical activation and heat treatment and to shed light on the structural and physicochemical properties of the resulting ferrites.
EXPERIMENTAL PART
Materials. The following materials were used as feedstock:
– cobalt oxalate CoC2O42 ∙ H2O; the CoC2O42 ∙ H2O mass fraction is equal to 99.5%, the remainder is Fe2O3, NiO, and CuO impurities; the initial cobalt oxalate consisted mainly of particles up to 25 μm in size;
– iron oxalate FeC2O42 ∙ H2O; the of FeC2O42 ∙ H2O mass fraction is equal to 99.0%, the remainder is Fe2O3, CaO, MgO, and K2O impurities; iron oxalate consisted of particles up to 100 μm in size.
Sample preparation procedure. The samples were prepared by two methods:
– solid-phase synthesis, by mixing the initial components taken in molar stoichiometric ratio to obtain cobalt ferrite CoFe2O4, and subsequent heat treatment in a muffle furnace in the temperature range of 300 – 1100°C;
– joint mechanical activation of a mixture of iron oxalate and cobalt oxalate, taken in stoichiometric ratio to obtain cobalt ferrite, for 45 min and subsequent heat treatment of the resulting mixture in the temperature range of 300 – 450°C; machining was conducting in a VM-4 roll-ring vibratory mill (Czech Republic); the milling chamber diameter is equal to 98 mm and the total volume of the chamber 0.302 L, the oscillation frequency is equal to 930 min – 1 and the amplitude 10 mm; the mass of grinding media is equal to 1194 g, the mass of the loaded material 100 g, and the milling time 45 min.
Procedures. The chemical and physical properties of the initial and obtained components were studied by the following methods:
– x-ray phase analysis; x-ray diffraction patterns of the powders were obtained on a DRON-3M x-ray diffractometer (CuKá radiation, λ = 0.15406 nm, Ni filter); power supply parameters 40 kV and 20 mA; scanning speed 2 deg/min; initial slit 2 mm; detector slit 0.25 mm;
– thermogravimetry (TG) and differential scanning calorimetry (DSC) were conducted on an STA449 F3 Netzsch simultaneous thermal analyzer in an air atmosphere; the heating rate was equal to 5 K/min–1;
– surface area, adsorption-desorption isotherms, and pore size distribution data were obtained on a Sorbi-MS analyzer; the BET specific surface area was determined by means of low-temperature nitrogen adsorption-desorption; the adsorption-desorption isotherms were obtained by the dynamic method of low-temperature nitrogen adsorption-desorption of nitrogen; prior to the investigation the samples were dried in a nitrogen stream at 150°C for 60 min;
– scanning electron microscopy (SEM); the measurements were conducted with a Vega 3 TESCAN microscope.
Methods of calculation. Interplanar spacing d, coherent scattering zone (CSZ) size, and rms microstrains ε were calculated from the XRD data.
The crystalline phases in the diffraction patterns were identified by comparing the calculated interplanar distances with the interplanar distances given in the ASTM database. Interplanar distances are calculated according to the Bragg equation [2]
where λ is the wavelength; Θ = Xc /2 is the diffraction angle, which was calculated from the position of the reflection center of gravity [3]:
where I is the intensity at the diffraction angle θ and Imax is the maximum intensity.
The size of the coherent scattering zone (CSZ) was calculated using the modified Scherrer equation [4], which after linearization has the form
where DSCR is the size of the coherent scattering zone; ε is the value of root-mean-square microstrains (MS); βph is the integral physical broadening of the x-ray profile of the sample, which was calculated using the Gaussian function [3]
where \( {\upbeta}_{\mathrm{s}}^2 \) is the integral broadening of the x-ray profile by the sample; \( {\upbeta}_{\mathrm{inst}}^2 \) is the instrumental broadening. The broadening of the x-ray profile of the standard was used as instrumental broadening.
The integral broadening of the x-ray profile was calculated according to the equation [13]
The interplanar distances and the corresponding Miller indices were used to calculate the unit cell parameters. For the cubic system, the equation has the form [14]
The specific energy E was calculated by the method proposed in [2]. Specific energy is only the net energy that is applied from the grinding media to the powder. This value does not include energy losses in the various mechanical components of the mill equipment.
RESULTS AND DISCUSSION
It was found by means of x-ray phase analysis that the mechanical activation (MA) process is attended for 1 – 45 min by a gradual decrease in the integral intensity of the reflections of iron oxalate phases, which indicates its gradual amorphization. It was established that in the course of mechanical activation oxalate crystallites decrease in size from 195 Å for the initial FeC2O4 ∙ 2H2O and to 88 Å after 45 min of MA, while the microstrains increase from 0.1 to 0.82% (Table 1). The fact that an CSZ is smaller than a crystallite, since it does not include external amorphous layers of the crystallite, and the magnitude of microstrains includes all crystal lattice distortions caused by point (Frenkel and Schottky defects), linear (edge and screw dislocations), and other types of defects. Cobalt oxide reflections are not observed in the x-ray pattern, which indicates that it is in the x-ray amorphous state. Formations of new phases were not recorded at the mechanical activation stage.
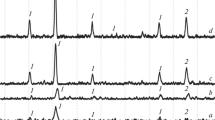
The calcination of a mixture subjected to mechanical activation at 300°C leads to the appearance of the γ-Fe2O3 phase, as evidenced by x-ray phase analysis (Fig. 1a ), an increase in the calcination temperature to 350°C (Fig. 1b ) does not lead to a significant change in radiographs. A subsequent increase in temperature to 400 – 450°C leads to the appearance of a set of reflections characteristic of cobalt ferrite and its subsequent crystallization (Fig. 1c ).

X-ray patterns of the products of mechanochemical activation of FeC2O4 ∙ 2H2O and CoFe2O4 ∙ 2H2O heat-treated at different temperatures: a) 300°C; b ) 350°C; c) 400°C; d ) 450°C; 1 ) γ-Fe2O3;2) CoFe2O4.
X-ray diffraction patterns of calcined samples obtained by solid-phase synthesis without preliminary mechanochemical treatment are displayed in Fig. 2a – d. In this case, calcination at 300°C leads to the formation of an x-ray amorphous mixture of iron and cobalt oxides resulting from the decomposition of iron and cobalt oxalates (Fig. 2a ). Heat treatment at 350°C leads to the appearance of reflections characteristic of γ-Fe2O3, which indicates its crystallization (Fig. 2b ). Raising the temperature to 450°C leads to the transition of the γ-Fe2O3 phase to α-Fe2O3 and the appearance of a crystalline phase of cobalt oxide Co3O4 (Fig. 2c ). Subsequent calcination at 450 – 850°C leads only to the crystallization of iron and cobalt oxides (Fig. 2d ). The formation of single-phase cobalt ferrite is noted only at 1100°C (Fig. 2b ).

X-ray diffraction patterns of the products of heat treatment of a mixture of FeC2O4 ∙ 2H2O and CoFe2O4 ∙ 2H2O, obtained at different temperatures: a) 300°C; b ) 350°C; c) 450°C; d ) 500°C; e) 850°C; f ) 1100°C; 1 ) γ-Fe2O3; 2 ) α-Fe2O3; 3) Co3O4; 4 ) CoFe2O4.
The calcination process in a sample obtained by solid-phase synthesis without preliminary mechanochemical treatment is accompanied by six thermal effects (Fig. 3a ):

DSC and TG curves for a mixture of FeC2O4 ∙ 2H2O and CoFe2O4 ∙ 2H2O without pretreatment (a) and 45 min after mechanical activation (b ).
I — endothermic effect occurring at temperature 130 – 215°C is associated with the removal of crystal water;
II, III — exothermic effects at temperatures 215 – 255 and 265 – 295°C are due to the decomposition of iron and cobalt oxalates, respectively, as well as simultaneously occurring carbon monoxide oxidation processes:
IV—exothermic thermal effect occurring at temperature 350 – 410°C is due to the transition of γ-Fe2O3 to α-Fe2O3;
V — endothermic effect observed at 900 – 940°C is due to the decomposition of cobalt oxide Co3O4 with oxygen release:
The TG curve of the given sample (Fig. 4a ) clearly depicts stepped losses of mass due to the removal of crystal water, decomposition of oxalates, and oxygen emission from the Co3O4 structure.

SEM image of cobalt ferrite at different synthesis temperatures: a) 350°C; b ) 450°C; c) 1100°C.
The mechanochemical activation of the oxalate mixture effects a change in the shape of the DSC curve (see Fig. 3b ). The thermic effects of the decomposition of iron oxalates I' and cobalt II' are shifted by 8 and 12°C, respectively, towards lower temperatures. The TG curve also shows stepwise weight loss but the curve is flatter. The thermic effect IV' at temperature 300 – 525°C is effected by ongoing processes of crystallization of cobalt ferrite CoFe2O4. The endothermic effect at 650 – 900°C is due to the emission of oxygen from the CoFe2O4 structure, which is additionally evidenced by the mass loss observed on the TG curve (see Fig. 3b ). Evidently, on joint mechanical activation of the initial components not only their comminution and intensive mixing occur, in the course of which the components are ground on top of one another other, but in addition excess energy accumulates in the form of defects in the crystal structure, which helps to lower the decomposition temperature of the initial components and the formation temperature of the final product— cobalt ferrite.
Investigation of cobalt ferrite samples by means of scanning electron microscopy (see Fig. 4a – c) shows that the samples obtained by means of mechanochemical synthesis consist of spherical particles 0.1 μm in size, which in turn form larger aggregates 5 – 25 μm in size (see Fig. 4a). An increase in the calcination temperature effects an increase in the size of the particles themselves as well as of the aggregates formed from them (see Fig. 4b ). Cobalt ferrite, obtained by solid-phase interaction of the initial components without preliminary mechanochemical activation (see Fig. 4c), consists of spherical particles with a clearly defined morphology. The particles of this sample are spherical with size 2.5 – 5.0 μm. The high heat treatment temperature (1100°C) leads to the sintering of particles into larger aggregates (see Fig. 4c).
The characteristics of cobalt ferrites obtained by various methods are summarized in Table 2.
Cobalt ferrite possesses a cubic crystal lattice in which, according to the database, a = b = c = 8.391 Å. The calculated parameters for the crystal lattice of cobalt ferrite obtained by solid-phase synthesis without preliminary mechanical activation at temperature 1100°C coincide with the published data (see Table 2). Cobalt ferrite obtained by solid-phase synthesis with preliminary mechanical activation of the initial components has somewhat distorted crystal lattice parameters (see Table 2).
It is evident that mechanical activation effects the formation of cobalt ferrite with a defective structure, which is also evidence by calculations of root-mean-square microstrains, whose values in the samples obtained using mechanochemical synthesis is 2 – 3 times greater than in a sample obtained by the customary solid-phase synthesis (see Table 2).
Two important characteristics of catalytic systems are their specific surface area and porosity. An increase in the failure temperature of samples effects a decrease in their specific surface area and total pore volume (see Table 2). Figure 5a – c show nitrogen adsorption-desorption isotherms of samples obtained by means of mechanical activation. These are type IV isotherms; such pores are characteristic for bodies containing predominantly transition pores (mesopores) in Dubinin’s classification, i.e. pores with diameters tens to hundreds of angstroms.

Nitrogen adsorption-desorption isotherms on cobalt ferrite samples and corresponding pore size distributions. Heat treatment temperature, °C: a) 300; b ) 400; c) 450; V, ml NDT/g — volume of desorbed gas at normal desorption temperature, related to the predetermined mass of a sample.
The sample calcined at 350°C has only mesopores with average diameter 3.5 to 29.4 nm, with 71.92% of the diameters ranging from 5.9 to 14.9 nm (Fig. 5a ). Raising the calcination temperature of the samples to 400 – 450°C effects an increase in the content of larger mesopores and the appearance of macropores (Fig. 5b and c). For example, a sample calcined at 400°C has pores with diameter 15.0 to 43.6 nm, which diameter 57.29% of all pores have (Fig. 5b ). As the calcination temperature increases, the specific surface area and total pore volume decrease (see Table 2), which is associated with gradual sintering of the samples, which results in the coarsening of particles and the formation of larger and tightly interconnected aggregates from them. It was established that the sample obtained without mechanochemical treatment has a very low specific surface area equal to 0.6 m2/g and is non-porous. This is also confirmed by SEM images of this sample, showing that the powder particles have a smooth surface and coalesce into dense aggregates without pores (Fig. 5c ).
CONCLUSIONS
Preliminary mechanochemical treatment of the initial components makes it possible to lower the synthesis temperature of single-phase cobalt ferrite by three-fold in comparison with the solid-phase interaction of the initial components not subjected to preliminary activation. Since the formation of new phases does not occur as a result of mechanochemical activation, it is obvious that the main channel for the relaxation of the supplied energy is the accumulation of defects in the structure of iron and cobalt oxalates, which in turn effects the intensification of their decomposition processes. The high homogeneity of the system subjected to preliminary mechanochemical activation contributes to a more complete and intense interaction of the resulting iron and cobalt oxides, which can also be the reason for lowering the synthesis temperature of the final product. Cobalt ferrite obtained by means of mechanochemical synthesis has a more developed specific surface and porous structure, which is explained by its milder conditions of synthesis.
This work was performed within the framework of the State mission for the implementation of R&D and with the support of a President of the Russian Federation scholarship. Topics No. FZZW-2020-0010 and No. 15493GU/2020. The research was performed using the resources of the Center for the Collective Use of Scientific Equipment of the ISUCT (with the support of the Russian Ministry of Education and Science, agreement No. 075-15-2021-671)
References
C. Kaçar, B. Dalkiran, P. E. Erden, and E. Kiliç, “An amperometric hydrogen peroxide biosensor based on Co3O4 nanoparticles and multiwalled carbon nanotube modified glassy carbon electrode,” J. Appl. Surf. Sci., 311, 139 – 146 (2014). DOI: https://doi.org/10.1016/j.msec.2011.10.028
F. A. Tourinho, R. Franck, and R. Massart, “Aqueous ferrofluids based on manganese and cobalt ferrites,” J. Mater. Sci., 25(7), 3249 – 3254 (1990).
V. Cabuil, V. Dupuis, D. Talbot, and S. Neveu, “Ionic magnetic fluid based on cobalt ferrite nanoparticles: influence of hydrothermal treatment on the nanoparticle size,” J. Magn. Magn. Mater., 323(10), 1238 – 1241 (2011). DOI: https://doi.org/10.1016/j.jmmm.2010.11.013
J. B. Silva, C. F. Diniz, R. M. Lago, and N. D. Mohallem, “Catalytic properties of nanocomposites based on cobalt ferrites dispersed in sol-gel silica,” J. Non-Cryst. Solids, 348, 201 – 204 (2004).
A. A. Thomas, A. Pietro, M. Emanuela, et al., “Synthesis and characterization of copper ferrite magnetic nanoparticles by hydrothermal route,” J. Chem. Eng. Trans., 47, 151 – 156 (2016). DOI: https://doi.org/10.3303/CET1647026
Z. Zi, Y. Sun, X. Zhu, and W. Song, “Synthesis and magnetic properties of CoFe2O4 ferrite nanoparticles,” J. Magn. Magn. Mater., 321, 1251 – 1256 (2009).
V. Gopalan, P. A. Joy, I. A. Al-Omari, et al., “On the structural, magnetic and electrical properties of sol–gel derived nanosized cobalt ferrite,” J. Alloys Compd., 485 711 – 718 (2009).
T. P. Braga, B. M. C. Sales, A. N. Pinheiro, et al., “Catalytic properties of cobalt and nickel ferrites dispersed in mesoporous silicon oxide for ethylbenzene dehydrogenation with CO2,” J. Catal. Sci. Technol., 1(8), 1383 – 1392 (2011). DOI: https://doi.org/10.1039/C1CY00176K
A. A. Magaeva, E. P. Naiden, O. G. Terekhova, et al., “Mechanochemical synthesis, phase composition, structural parameters, and magnetic properties of manganese ferrospinels,” J. Nanotechnologies in Russia, 8(7 – 8), 495 – 501 (2013). DOI: https://doi.org/10.1134/S1995078013040083
L. J. Berchmans, R. Karthikeyan, M. Helan, et al., “Mechanochemical synthesis and electrochemical characterization of nano crystalline calcium ferrite,” J. Catal. Lett., 141(10), 1451 – 1457 (2011). DOI: https://doi.org/10.1007/s10562-011-0636-9
E. Manova, D. Paneva, B. Kunev, et al., “Mechanochemical synthesis and characterization of nanodimensional iron-cobalt spinel oxides,” J. Alloys Compd., 485(1 – 2), 356 – 361 (2009). DOI: https://doi.org/10.1016/j.jallcom.2009.05.107
K. O. Denisova, A. A. Ilyin, R. N. Rumyantsev, et al., “Low-temperature synthesis and catalytic activity of cobalt ferrite in nitrous oxide (N2O) decomposition reaction,” J. Catalysts, 11(8), 889 (2021). DOI: https://doi.org/10.3390/catal11080889
T. Ekström, C. Chatfield, W. Wruss, M. Maly-Schreiber, “The use of x-ray diffraction peak-broadening analysis to characterize ground Al2O3 powders,” J. Mater. Sci., 20(4), 1266 – 1274 (1985).
H. Heegn, “On the connection between ultrafine grinding and mechanical activation of minerals,” J. Aufbereitungs-technik, 30(10), 635 – 642 (1989).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Steklo i Keramika, No. 1, pp. 21 – 30, January, 2022.
Rights and permissions
About this article

Cite this article
Ptitsyna, K.O., Il’in, A.A., Rumyantsev, R.N. et al. Mechanochemical and Ceramic Synthesis of Cobalt Ferrite. Glass Ceram 79, 15–21 (2022). https://doi.org/10.1007/s10717-022-00446-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10717-022-00446-9