
Abstract
Podocarpus sellowii (Podocarpaceae) is one of only a few gymnosperms native to Brazil and the sole species of the genus found in the northeastern region of that country. It has a very restricted distribution in this region, with only three known populations in highland forests (called Brejos de Altitude), which apparently have been isolated from each other since the Pleistocene. Due to this long-term isolation and the fact that these populations have few adult individuals and suffer great anthropogenic pressure, low genetic variability is expected, compromising their long-term viability. The present work assessed the genetic variability and structure of northeastern populations of P. sellowii to investigate the role of Pleistocene glaciations on the genetic relationships between them and to propose strategies for their conservation by analyzing the SSR and ISSR markers of adult and juvenile individuals. Low genetic diversity was found with both markers, associated with a high differentiation of the Brejo de Baturité population in relation to the others—suggesting their isolation at different points in time, probably during the Pleistocene. Actions directed towards increasing the genetic diversity of these populations will be needed, such as planting seedlings with high genetic variability—but the high degrees of differentiation observed between the populations must be taken into account.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The classical Pleistocene refuge hypothesis (Haffer 1969; Vanzolini and Williams 1970) has been invoked for many years as the main explanation for the high rates of endemism and interspecific diversity observed in South American forests (Whitmore and Prance 1987; Carnaval and Moritz 2008). Recent phylogeographic studies have shown that refuges have played important roles in the diversification of the Atlantic rainforest (Cabanne et al. 2008; Carnaval et al. 2009; Maldonado-Coelho 2012), although their importance to the Amazon forest remains controversial (Hoorn et al. 2010; Ribas et al. 2012; Thomas et al. 2012). Brejos de Altitude (hereafter referred to as Brejos) are relict enclave rainforests in Caatinga (dryland) vegetation highlands, and are considered to be biogeographical sub-regions of the Atlantic Forest and forest refuges within that xeric landscape (Andrade-Lima 1982; da Silva and Casteleti 2003). Due to their distinct climates in relation to the surrounding Caatinga vegetation and their long geographic isolation from other Atlantic Forest remnants (probably since the Pleistocene glaciations), these areas exhibit high levels of endemism (Félix and Carvalho 2002; Siqueira-Filho 2002). However, due to their distinct highland climates in relation to the surrounding dry Caatinga, these areas are well suited for agricultural use and have therefore been largely degraded, making them the most threatened portions of the Atlantic Forest domain (Tabarelli and Santos 2004; Oliveira et al. 2007).
The few published phylogeographic studies of Brejo populations have revealed moderate to high levels of population differentiation, usually correlated with isolation due to Pleistocene climatic fluctuations. Brejo populations localized in the northernmost portion of their distribution range are believed to have survived the driest periods of the Pleistocene (Carnaval and Bates 2007), and were possibly connected to both the Amazon and Atlantic Forests during cycles of forest expansion (Andrade et al. 2007). Different levels of genetic diversity have been observed in Brejo populations. Low levels of genetic variability were observed in enclave populations of two frog species (Carnaval and Bates 2007), while no reductions in the levels of genetic diversity were observed in the plant Anthurium sinuatum (Andrade et al. 2009). According to the latter authors, this might indirectly support the idea that those relict areas were only recently formed (c. 5,000 years BP) by the contraction of a formerly more widespread, forested coast connected with the Amazonia forest.
Among the plant species found in Brejos sites, Podocarpus sellowii Klotzsch ex Endl. (Podocarpaceae) stands out, with Podocarpus being the largest genus of gymnosperm in the southern hemisphere. Two species of this genus have been reported as being well distributed in Brazil: P. lambertii, found in the meridional region of the country with a cooler climate; and P. sellowii, found both in warm and cold regions, but with sparser distribution (Lima and Barbosa 1998). P. sellowii is the only conifer native to northeastern Brazil (Lima and Barbosa 1998), with only four known populations reported for the states of Pernambuco (Andrade-Lima 1971), Ceará (Figueiredo et al. 1990), Sergipe and Bahia (Duarte 1973). It is not clear, however, if the population from Bahia is that of P. sellowii, as it exhibits some morphological characters more closely associated with P. lambertii (L. Félix, unpublished results). Only small numbers of adult individuals comprise the Pernambuco and Ceará populations (Lima and Barbosa 1998; Araújo et al. 2007). Together with the long-term geographical isolation of these Brejos areas, genetic isolation might have reduced the genetic variability of this dioecious (and therefore exclusively xenogamic) species (Aguilar et al. 2008). A positive correlation between genetic and geographic distances has been observed in another species of this genus, P. parlatorei (which is characteristic of the montane Yungas forests growing along the eastern slopes of the Andes Mountains and occurring in disjunct populations in Argentina and Bolivia), which has been attributed to the glacial events of the Pleistocene (Quiroga and Premoli 2007).
Populations of P. sellowii found in the Atlantic Forest in Minas Gerais State in southeastern Brazil (which have large numbers of individuals and are not presently considered threatened) were recently analyzed using isozyme markers, and revealed little differentiation among the eight subpopulations analyzed, with high values of genetic diversity (Gonçalves 2008). Since the northeastern populations of P. sellowii have reduced sizes (Lima and Barbosa 1998; Araújo et al. 2007) and are geographically isolated, their genetic statuses should be evaluated and taken into account when formulating strategies for their conservation. Thus, the present work analyzed the genetic diversity and population structures of three northeastern populations of P. sellowii to shed light on the role of Pleistocene glaciations and the subsequent isolation of Brejo populations, and to provide information for the conservation of this species.
Materials and methods
Plant sampling and DNA extraction
Leaves were collected from adult and juvenile (at least 1 m high, but non-reproductive) individuals from three populations of P. sellowii (Fig. 1a): leaves from 15 juveniles and 15 adults individuals within a population (BT) located on private property between the counties of Guaramiranga and Pacotí, in the Maciço do Baturité (Ceará State, 04°15′46″S × 38°55′58″W); from 15 juveniles and 20 adults in a second population at Brejo dos Cavalos (BC) in the Professor Vasconcelos Sobrinho Ecological Reserve in the city of Caruaru (Pernambuco State, 08°22′09″S × 36°05′00″W); and from 15 juveniles and 20 adults from a third population (IT) in the Serra da Itabaiana National Park (Sergipe State, 10°40′52″S × 37º25′15″W). All of the leaves were dried and stored over silica gel (Chase and Hills 1991) until assayed.

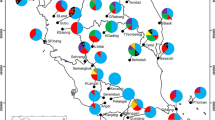
Sampling sites of Podocarpus sellowii populations (a) and the distribution of genetic diversity using SSR and ISSR markers (b–f). a Map of Brazil, indicating the locations of the populations sampled: Baturité (BT, red), Brejo dos Cavalos (BC, blue), and Itabaiana (IT, green); b NJ phenogram based on SSR data; c UPGMA dendrogram based on D (according to Nei 1972) using SSR data; d NJ phenogram based on ISSR data; e UPGMA dendrogram based on D (according to Nei 1972) using ISSR data; f Clustering of individuals based on STRUCTURE analysis based on SSR data
DNA was extracted following the protocol described by Doyle and Doyle (1987) (with minor modifications for time optimization as described in the supplementary file), or using the commercial Plant Genomics DNA Mini Kit YGP100 (Real Biotech Corporation) following the manufacturer’s instructions. For SSR library construction, DNA was extracted (according to Zellinger et al. 2007) from the fresh leaves of one individual cultivated in the experimental garden of the Laboratory of Plant Cytogenetics and Evolution at the Federal University of Pernambuco—UFPE. All DNA samples were quantified in 1 % agarose gel stained with ethidium bromide (0.001 mg/mL), and the gels visualized using a UV transilluminator coupled to a Doc-Print-II gel documentation system (Vilber Lourmat).
SSR markers
The construction of a microsatellite-enriched genomic DNA library for (GT)8 and (CT)8 repeats followed the protocol described by Mori et al. (2010). Ninety-five SSR-enriched clones were sequenced in both directions with T7 and SP6 primers using the Big Dye® Terminator Cycle Sequencing Kit v3 (PerkinElmer Applied Biosystems) in a ABI PRISM® 377 automatic sequencer. Sequences were aligned and edited using Geneious software. Microsatellites were identified with the help of Gramene (Youens-Clark et al. 2011). The primers were designed using FastPCR v. 3.6.97 (Institute of Biotechnology, University of Helsinki, Finland) and Primer3 v. 0.4.0 (http://primer3.sourceforge.net/), while the absence of dimers and hairpins was checked with NetPrimer (http://www.premierbiosoft.com/netprimer/). The designed primers (Table S1) were initially tested using the DNA of the same individual from which the SSR-enriched library was established, whereas those with satisfactory amplifications were analyzed in the remaining individuals. Amplifications were carried out in 25 µL reaction volumes containing 6 ng of genomic DNA, 1.25 U of Taq DNA polymerase (GE), 1× PCR buffer (100 mM Tris–HCl pH 9.0, 15 mM MgCl2 and 500 mM KCl), 0.16 mM of each dNTP (GE), and 0.8 µM of each primer. All amplifications were performed in a Mastercycler thermocycler (Eppendorf). PCR amplifications were composed of one initial denaturing step at 94 °C for 1 min, followed by 30 cycles of 1 min at 94 °C, 1 min at 53–60 °C, and 1 min at 72 °C, with a final extension of 5 min at 72 °C. The PCR products were initially analyzed in 3 % agarose in 1× TAE buffer gels stained with ethidium bromide; those with satisfactory amplifications were genotyped in 6 % polyacrylamide in 1× TBE buffer gels with a 10 bp ladder to determine product sizes (Invitrogen, CA, USA). The DNA fragments were visualized by silver staining, following Creste et al. (2001).
ISSR markers
A set of 28 primers (UBC set no. 9) developed by the Biotechnology Laboratory of the University of British Columbia were tested in a subset of individuals from different populations; the 14 primers found to be more polymorphic were used for genotyping fifteen juveniles per population. One individual from the BC population (BC J3) was excluded from the analysis due to poor amplification. The primers were 17–18 nucleotides long and contained di-, tri- or tetranucleotides anchored with one, two or three nucleotides (Table S2).
Amplifications were performed according to the protocol described by Bornet and Branchard (2001), with the following modifications: the reactions were carried out in 20 µL volumes containing 4 ng of genomic DNA, 1 U of Taq DNA polymerase (Fermentas), 1x PCR buffer (100 mM Tris–HCl pH 8.8, 0.8 % Nonidet P40 and 500 mM KCl), 2 mM MgCl2, 0.25 mM of each dNTP (GE), and 0.5 µM of primer. All amplifications were performed in a Mastercycler thermocycler (Eppendorf). PCR amplification consisted of one initial denaturing step at 94 °C for 5 min, followed by 25 cycles of 45 s at 94 °C, 1 min at 50.4–58 °C, 30 s at 72 °C, with a final extension of 15 min at 72 °C. The PCR products were separated in 1.5 % agarose in 1× TAE buffer gels and stained with ethidium bromide.
Data analysis
For each SSR locus, the number of alleles, total expected heterozygosity (H T), expected heterozygosity within populations (H S), and observed heterozygosity (H O) (Nei 1987) were calculated using Fstat v. 2.9.3.2 software (Goudet 2001), in addition to Wright’s F statistics (F ST, F IS e F IT), and the R ST (non-weighted) and G ST genetic differentiation coefficients between populations. F ST indicates the degree of genetic differentiation between populations by the infinite alleles model, whereas R ST uses a stepwise model. G ST allows better comparisons between loci with different levels of genetic differentiation (Hedrick 2005). Deviations from the Hardy–Weinberg equilibrium were tested for each locus using GENEPOP 4.2 software (Raymond and Rousset 1995).
We also assessed the potential impact of mutation versus drift on the detected genetic patterns by the permutation of allele sizes using SPAGeDi 1.4 software (Hardy and Vekemans 2002). Permutation tests were applied to test the null hypothesis that population differentiation is not affected by differences in allele sizes (Hardy et al. 2003). Potential historical signals were thus assessed by permuting allele sizes at each microsatellite locus among allelic states (20,000 replicates) to simulate the distribution of R ST values (pR ST) with 95 % confidence intervals (CI). These p values (pR ST) indicate whether there are significant differences between permuted R ST values compared to global R ST values.
For the ISSR markers, clear amplification products were scored as present (1) or absent (0). Fragments that were present in less than 5 % or more than 95 % of the individuals were considered monomorphic, following the criteria suggested by Ott (1992).
A dissimilarity matrix was created for both marker types using the Jaccard index (Perrier et al. 2003) and was subsequently used to generate a phenogram based on the Weighted Neighbor Joining algorithm (bootstrap of 1,000 replicates), using DARwin 5.0 software (Perrier and Jacquemoud-Collet 2006). TFPGA v. 1.3 software (Miller 1997) was used to estimate a genetic distance matrix (Nei 1972), which was used to generate a UPGMA dendrogram. Additionally, gene diversity (H E ) was computed for each population, and the distribution of molecular variation within one hierarchical level as measured by Analysis of Molecular Variance (AMOVA), as well as the total and pairwise F ST were calculated using Arlequin 3.5 software (Excoffier and Lischer 2010). GENEPOP 4.2 software (Raymond and Rousset 1995) was also used to compute the number of migrants (Nm) for the SSR data. The population structure was tested using the SSR data and STRUCTURE v 2.3.3 software (Pritchard et al. 2000), with 10 replicates for each K value (which ranged from 1 to 5). Each run consisted of a burn-in period of 10,000 with one million MCMC replications after burn-in. The inference of K was evaluated using the ∆K model (Evanno et al. 2005).
Results
Selection and polymorphism of the molecular markers
The ninety-five sequenced clones obtained from the microsatellite-enriched P. sellowii library were assembled in 57 contigs, suggesting some redundancy among the sequences. Thirty of them had at least one microsatellite, with a total of 45 microsatellite regions. Even though the library had been enriched for the dinucleotides (GT)8 and (CT)8, several other microsatellites were identified, including tetra and pentanucleotides. Primers could be designed for 14 SSR regions. Eighteen primer pairs were tested for those regions, but only seven pairs showed satisfactory amplifications, with fragments having expected sizes (Table S1).
Three (C12_1, D4 and F2) out of the seven SSR loci were polymorphic in the populations analyzed, with an average of 2.33 alleles per polymorphic locus (Table 1). The proportions of polymorphic loci (P95) per population were equal in the three populations analyzed (0.2857), with the BC and IT populations showing polymorphism for the D4 and F2 loci, and the BT population for D4 and C12_1.
The 14 selected ISSR loci generated 83 clear fragments that were used for genotyping (Table S2). These fragments had between 300 bp and 2.5 kb, with 30 of them (36.15 %) being present in 5–95 % of the individuals, and were thus considered polymorphic. Fifty-three bands (63.85 %) were considered monomorphic; fifty-two of them were found in all individuals, whereas one was only found in a single individual.
Population variability
The total observed heterozygosity (H 0) for SSR loci was 0.393, ranging from 0.190 for C12_1 to 0.605 for F2. The average expected heterozygosity within populations (H S) was 0.301, ranging from 0.137 (C12_1 locus) to 0.437 (D4 locus) (Table 1). The average gene diversity (H E) was 0.302, ranging from 0.285 for the IT population to 0.323 for the BC population (Table 2). F IS values were negative for all populations, ranging from −0.101 for the BT population to –0.558 for the IT population, indicating an excess of heterozygotes. Even though polymorphism was low, the Hardy–Weinberg test was congruent with this result, indicating an excess of heterozygotes for the BT (p = 0.0028) and IT (p = 0.0000) populations, whereas the per locus test indicated excesses for the C12_1 (p = 0.0222) and F2 (p = 0.0000) loci. The values for the BC population were non-significant (p = 0.3543). Two private alleles were found in the BT population, one for C12_1 and one for D4 (Tables 1, 2).
The variability of the ISSR markers was also found to be low, with 16 loci being polymorphic in the BT and BC populations and 17 in IT. The BT population exhibited the lowest value for average H E (0.067), while the highest value was found for IT (0.077). Private bands were highest in the BT and lowest in the IT populations (Table 2).
Genetic structure
The genetic distance matrices computed from F ST and from the Nei diversity index (Nei 1972) obtained for the SSR loci indicated that the BC and IT populations were differentiated from BT (Tables 3 and S3), while the divergence between BC and IT was very low (or not significant) (Tables 3 and S3; Fig. 1c). With the ISSR loci, on the other hand, the pairwise F ST values indicated significant differentiation between all of the populations, although the difference between BC and IT was lower than between BT and BC or BT and IT (Table 3).
The results of the random permutation of different allele sizes among allelic states at each locus and multilocus revealed that R ST estimates were not significantly higher than pR ST (Table S3), suggesting that allele size is not informative for population differentiation.
The genetic distance matrix computed from the Nei genetic diversity index for ISSR markers showed that BT was highly divergent from the other two populations (Table S4). These population divergences can also be visualized in the Weighted Neighbor Joining phenograms (Fig. 1b, d), where the BT population was isolated from the others. For the ISSR loci (Fig. 1d), all individuals of a given population grouped together, although with two subpopulations within IT (which correlated with the two subpopulations sampled in that area). In general, the F ST fixation indexes were high for both makers, although most of the variation of the SSR markers was seen within, and not among, populations, as with the ISSR markers (0.259 for SSR and 0.633 for ISSR, Table 4). The number of migrants calculated using the SSR data after correcting for size was 0.037321. Simulations performed using STRUCTURE software consistently identified K = 2 clusters (Figure S1), with BT individuals grouped in one cluster; some analyzed individuals, however, showed admixed ancestry (Fig. 1f).
Discussion
Genetic diversity
The results from the SSR and ISSR markers were congruent, and indicated low genetic diversity in the northeastern populations of P. sellowii. Among the SSR and ISSR loci analyzed, 57 and 63.85 % were monomorphic, respectively. Similar low levels of variability have been reported for some gymnosperms, with Peakall et al. (2003) describing low levels of genetic diversity in three species of Araucariaceae—ranging from no polymorphism at any of the SSR loci analyzed in Wollemia nobilis and Agathis robusta to only five out of 21 SSRs being polymorphic in Araucaria cunninghamii. In a study with another species of Podocarpaceae, Dacrycarpus imbricatus (a species also suffering from habitat loss), a very high level of polymorphism was observed (96 %) using ISSR markers (Su et al. 2010). Although genetic erosion is not an obligatory consequence of habitat loss, low genetic variability was, in fact, observed for the small isolated populations studied here, indicating genetic erosion—possibly as a consequence of genetic drift.
Besides the low observed proportions of polymorphic loci, low numbers of alleles per loci were also seen at the SSR loci. The mean number of alleles found for P. sellowii (an obligate allogamous species) in the microsatellite analysis was closer to those found in autogamous and/or annual species (1.5–6.2) than those of allogamous evergreen species (5.5–27.6) (Hokanson et al. 1998). Additionally, the gene diversity values (H E) found for P. sellowii were lower than those commonly observed in other tropical tree species (Bizoux et al. 2009) or other conifers (Peakall et al. 2003; Rungis et al. 2004; Marquardt et al. 2007). The low polymorphism associated with both types of markers in P. sellowii indicated that the populations analyzed had low genetic variability—which could lead to increased vulnerability to external factors and increased negative effects of deleterious alleles.
The low genetic diversity observed in the northeastern populations of P. sellowii does not appear to be characteristic of the species (or even the genus). Gonçalves (2008) used allozyme markers to analyze three populations of P. sellowii from Minas Gerais State in southeastern Brazil judged not to be threatened and observed eight subpopulations, in accordance with natural geographic barriers. Genetic diversity values were high, even higher than those found for other gymnosperm species, and 100 % of the loci analyzed were polymorphic (unlike the present work). Additionally, when one individual from Minas Gerais and two individuals from Rio Grande do Sul (southern Brazil) were analyzed using polymorphic SSR markers, new alleles for the C12_1 and F2 loci were found (data not shown). This suggests that the low genetic variability observed in the northeastern populations of P. sellowii is most likely related to high levels of genetic drift due to the small sizes of the remnant populations at each Brejo site, and to low levels of gene flow between these populations (which have probably been isolated since the Pleistocene) (Behling et al. 2000).
Population structuring
Although we cannot exclude the possibility that the high observed differentiations between these small Brejo populations are solely due to genetic drift, we favor the long term isolation hypothesis. This high differentiation is particularly evident at the Brejo de Baturité site in comparison to the others, and is reflected in: the number of migrants (Nm) lower than 1; the presence of private alleles (more numerous in BT); the high levels of pairwise genetic differentiation (F ST) in all comparisons including this population; and the STRUCTURE results. The presumed stronger isolation of the Brejo de Baturité population is in accordance with previous studies (Carnaval and Bates 2007). This region is floristically more similar to the Amazon and the Atlantic Forest north of the São Francisco River than to the other northeastern Brejos sites (Santos et al. 2007). This pattern suggests that connections with the Baturité site were lost earlier than the connections between Brejo dos Cavalos and Itabaiana.
The strong similarity between the IT and BC populations suggests a recent loss of connectivity, or low levels of gene flow, allowing relictual cohesion among individuals. Interestingly, Silva (2011) encountered Coprophanaeus bellicosus (Coleoptera) at Brejo dos Cavalos, a species previously recorded only for the Atlantic Forest south of the São Francisco River. According to this author, this represents a relictual population with a previously more widespread distribution, providing further evidence of connections between the Brejos located in central/southernmost part of their distribution range and the Atlantic rainforest.
Podocarpus sellowii populations from Minas Gerais, on the other hand, exhibited low levels of differentiation correlated with geographic distances (Gonçalvez 2008). The greatest geographic distance was slightly more than two kilometers, which explains the low observed structuring; the populations analyzed in the present study were separated by at least 400 km (BC-IT: ±400 km; BC-BT: ±700 km; BT-IT: ±900 km). Another species of Podocarpus, P. parlatorei (native to Argentina, Bolivia and Peru), showed strong genetic structuring (detected through isozyme assays) between the different populations that would likewise have suffered expansion and retraction events during the Pleistocene (Quiroga and Premoli 2007). Climatic oscillations during the Pleistocene appear to have contributed in different ways to the distribution of genetic diversity among populations of P. elatus (which is widely distributed throughout Australia)—depending on the geographic locations of those populations (Mellik et al. 2012). Likewise for P. sellowii, Caatinga expansions (with subsequent range contractions of humid Brejos forests during the Pleistocene) (de Oliveira et al. 1999; Behling et al. 2000; Auler et al. 2004; Wang et al. 2004) likely contributed to the current distribution of genetic variability in northeastern populations.
Historical processes
According to pollen record studies undertaken by Ledru et al. (2007), P. sellowii populations in northeastern Brazil expanded rapidly (during only 50–100 years) approximately 16,000 years ago, with this rapid expansion indicating the previous existence of several small habitat patches suitable to the occurrence of this species, as well as several routes between them. Despite uncertainties in the paleoclimatic history of northeastern Brazil, P. sellowii populations located in Caatinga enclaves are believed to have been isolated since at least the end of the Pleistocene (Behling et al. 2000), after periods of connectivity between each other and with the Amazon and Atlantic rainforests (de Oliveira et al. 1999). Both genetic markers used here showed considerable levels of differentiation between the populations analyzed– although losses of connectivity between the different Brejos could have occurred at different times—as the BT population probably lost connectivity first, while the BC-IT connectivity was later reduced. These data are in accordance with previous results that suggested high differentiation of the populations in Ceará State (Andrade et al. 2007, 2009; Carnaval and Bates 2007; Santos et al. 2007)
Conservation implications
Low numbers of adult individuals of P. sellowii were found in the northeastern populations studied. A floristic inventory of six areas in the Brejo de Baturité site identified only 16 individuals of P. sellowii in one area, among a total of 4,732 sampled plants (Araújo et al. 2007)—making it a rare species (Lima-Verde and Gomes 2007). This situation reflects low recruitment, as already noted by Lima and Barbosa (1998) and Araújo et al. (2007), which might be a consequence of the grouped distribution of the seedlings around the mother trees, as was observed in the field (and previously reported by Lima and Barbosa 1998). Reductions in effective population sizes may be contributing to genetic drift and mortality, due to lethal and sublethal alleles. This situation will require attention, as the diversity levels of these populations might become further reduced over time. The low genetic diversity seen in these northeastern populations indicates the necessity of implementing measures that will increase population sizes through the protection and improvement of habitats, the reduction of existing threats, and by introducing genetically differentiated individuals to increase genetic variability. These measures are particularly important now because these populations are greatly threatened by habitat loss, fragmentation, and agricultural expansion (Tabarelli and Santos 2004). However, due to the high differentiation of the Baturité population, it should be treated as a genetically unique and substantially differentiated site. More intact populations, such as those found in national reserves in Minas Gerais State, could provide sources of genetic variability transferable to northeastern populations—but the differences between the Brejo dos Cavalos, Itabaiana and southeastern Atlantic Forest populations must first be examined using morphological and molecular markers to determine if variability can be introduced without the risk of outbreeding depression. Conservation and restoration efforts are particularly relevant in light of the fact that Brejos are important reservoirs of diversity and represent the only vehicles for rapid expansion of the Atlantic Forest in response to future climate changes (Ledru et al. 2007).
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188
Andrade IM, Mayo SJ, Van Der Berg C, Fay MF, Chester M, Lexer C, Kirkup D (2007) A preliminary study of genetic variation in populations of Monstera adansonii var. klotzschiana (Araceae) from North-East Brazil, estimated with AFLP molecular markers. Ann Bot 100:1143–1154
Andrade-Lima D (1971) Flora de Pernambuco. Podocarpaceae. XXI Congresso Nacional Sociedade Botânica do Brasil. Ciência e Cultura 23:337
Andrade-Lima D (1982) Present-day forest refuges in Northeastern Brazil. In: Prance GT (ed) Biological diversification in the tropics. Plenum Press, New York, pp 245–251
Araújo FS, Gomes VS, Silveira AP, Figueiredo MA, Oliveira RF, Bruno MMA, Lima-Verde LW, Silva EF, Otutumi AT, Ribeiro KA (2007) Efeito da variação topoclimática na fisionomia e estrutura da vegetação da serra de Baturité, Ceará. In: Oliveira TS, Araújo FS (eds) Diversidade de Conservação da biota na Serra de Baturité, Ceará. Edições UFC, Coelce, Fortaleza, pp 73–136
Auler AS, Wang X, Edwards RL, Cheng H, Cristalli PS, Smart PL, Richards DA (2004) Quaternary ecological and geomorphic changes associated with rainfall events in presently Semi-arid Northeastern Brazil. J Quat Sci 19:693–701
Behling H, Arz HW, Pätzold J, Wefer G (2000) Late Quaternary vegetational and climate dynamics in Northeastern Brazil, inferences from marine core GeoB 3104-1. Quat Sci Rev 19:981–994
Bizoux JP, Daïnou K, Bourland N, Hardy OJ, Heuertz M, Mahy G, Doucet JL (2009) Spatial genetic structure in Milicia excelsa (Moraceae) indicates extensive gene dispersal in a low-density wind-pollinated tropical tree. Mol Ecol 18:4398–4480
Bornet B, Branchard M (2001) Nonanchored inter simple sequence repeat (ISSR) markers: reproducible and specific tools for genome fingerprinting. Plant Mol Biol Rep 19:209–215
Cabanne GS, d’Horta FM, Sari EHR, Santos FR, Miyaki CY (2008) Nuclear and mitochondrial phylogeography of the Atlantic Forest endemic Xiphorhynchus fuscus (Aves: Dendrocolaptidae): biogeography and systematics implications. Mol Phylogenet Evol 49:760–773
Carnaval AC, Bates JM (2007) Amphibian DNA shows marked genetic structure and tracks Pleistocene climate change in Northeastern Brazil. Evolution 61–12:2942–2957
Carnaval AC, Moritz C (2008) Historical climate modelling predicts patterns of current biodiversity in the Brazilian Atlantic forest. J Biogeogr 35:1187–1201
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science 323:785–789
Chase MW, Hills HH (1991) Silica gel: an ideal material for field preservation of leaf samples for DNA studies. Taxon 40:215–220
Creste S, Tulmann Neto A, Figueira A (2001) Detection of simple sequence repeat polymorphisms in denaturing polyacrylamide sequencing gels by silver staining. Plant Mol Biol Rep 19:299–306
da Silva JMC, Castelletti CHM (2003) Status of the biodiversity of the Atlantic Forest of Brazil. In: Galindo-Leal C, Câmara IG (eds) The Atlantic Forest of South America: biodiversity status, threats, and outlook. Center for Applied Biodiversity Science and Island Press, Washington, DC, pp 43–59
de Oliveira PE, Barreto AMF, Suguio K (1999) Late Pleistocene/Holocene climatic and vegetational history of the Brazilian caatinga: the fossil dunes of the middle São Francisco River. Palaeogeogr Palaeoclimatol Palaeoecol 152:319–337
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Félix LP, Carvalho R (2002) Diversidade de Orquídeas no Estado de Pernambuco. In: Tabarelli M, Silva JMC (eds) Diagnóstico da biodiversidade de Pernambuco, vol 1. Massangana, Recife, pp 207–217
Figueiredo MA, Fernandes A, Nepomuceno VAG (1990) Refúgio florístico de Podocarpus na Serra de Baturité—Ceará. XIV Reunião Nordestina de Botânica, Recife
Gonçalves FR (2008) Estrutura genética em populações naturais de Podocarpus sellowii Klotzsch (Podocarpaceae) na região do Alto Rio Grande, Sul de Minas Gerais. Dissertação de mestrado. Lavras: UFLA, p 72
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www.unil.ch/izea/softwares/fstat.html. Updated from Goudet (1995)
Haffer J (1969) Speciation in Amazonian forest birds. Science 165:131–137
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hardy OJ, Charbonnel N, Fréville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Hokanson SC, Szewc-McFadden AK, Lamboy WF, McFerson JR (1998) Microsatellite (SSR) markers reveal genetic identities, genetic diversity and relationships in a Malus x domestica borkh. core subset collection. Theor Appl Genet 97:671–683
Hoorn C, Wesselingh FP, ter Steege H, Bermudez MA, Mora A, Sevink J, Sanmartín I, Sanchez-Meseguer A, Anderson CL, Figueiredo JP, Jaramillo C, Riff D, Negri FR, Hooghiemstra H, Lundberg J, Stadler T, Säarkinen T, Antonelli A (2010) Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330:927–931
Ledru MP, Salatino MLF, Ceccantini G, Salatino A, Pinheiro F, Pintaud JC (2007) Regional assessment of the impact of climatic change on the distribution of a tropical conifer in the lowlands of South America. Divers Distrib 13:761–777
Lima TV, Barbosa DCA (1998) Levantamento da distribuição espacial de plantas jovens não estabelecidas e estabelecidas de Podocarpus sellowii Klotzsch, em Serra dos Cavalos, Caruaru—PE. Trabalho técnico, Imprensa Universitária, UFRPE, Recife, p 32
Lima-Verde LW, Gomes VS (2007) Plantas nativas da serra de Baturité, Ceará, com potencial ornamental. In: Oliveira TS, Araújo FS (eds) Diversidade de Conservação da biota na Serra de Baturité, Ceará. Edições UFC, Coelce, Fortaleza, pp 295–315
Maldonado-Coelho M (2012) Climatic oscillations shape the phylogeographical structure of Atlantic Forest fire-eyes (Aves: Thamnophilidae). Biol J Linn Soc 105:900–924
Marquardt PE, Echt CS, Epperson BK, Pubanz DM (2007) Genetic structure, diversity, and inbreeding of eastern white pine under different management conditions. Can J For Res 37:2652–2662
Mellik R, Lowe A, Allen C, Hill RS, Rossetto M (2012) Palaeodistribution modeling and genetic evidence highlight differential post-glacial range shifts of a rain forest conifer distributed across a latitudinal gradient. J Biogeogr. doi:10.1111/j.1365-2699.2012.02747.x
Miller MP (1997) Tools for population genetic analysis (TFPGA) 1.3: a Windows program for the analysis of allozyme and molecular population genetic data. Computer software distributed by author
Mori GM, Zucchi MI, Sampaio I, Souza AP (2010) Microsatellites for the mangrove tree Avicenia germinans (Acanthaceae): tools for hybridization and mating system studies. Am J Bot 97:e79–e81
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Oliveira TS, Figueiredo MA, Nogueira RS, Sousa SC, Souza SSG, Romero RE, Tavares RC (2007) Histórico dos impactos antrópicos e aspectos geoambientais da serra de Baturité, Ceará. In: Oliveira TS, Araújo FS (eds) Diversidade de Conservação da biota na Serra de Baturité, Ceará. Edições UFC; COELCE, Fortaleza, pp 21–27
Ott J (1992) Strategies for characterizing highly polymorphic marker in human gene mapping. AJHG 51:283–290
Peakall R, Ebert D, Scott LJ, Meagher PF, Offord CA (2003) Comparative genetic study confirms exceptionally low genetic variation in the ancient and endangered relictual conifer, Wollemia nobilis (Araucariaceae). Mol Ecol 12:2331–2343
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. http://darwin.cirad.fr/darwin. Acesso em: 30 Jun 2006
Perrier X, Flori A, Bonnot F (2003) Data Analysis Methods. In: Hamon P, Seguin M, Perrier X, Glasmann JC (eds) Genetic diversity of cultivated tropical plants. Enfield Science Publishers, Montpellier, pp 43–76
Pritchard JK, Stephens P, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quiroga MP, Premoli AC (2007) Genetic patterns in Podocarpus parlatorei reveal the long-term persistence of cold-tolerant elements in the southern Yungas. J Biogeogr 34:447–455
Raymond R, Rousset F (1995) GENEPOP version 1.2: population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Ribas CC, Aleixo A, Nogueira ACR, Miyaki CY, Cracraft J (2012) A paleobiogeographic model for biotic diversification within Amazonia over the past three million years. Proc R Soc B 279:681–689
Rungis D, Bérubé Y, Zhang J, Ralph S, Ritland CE, Ellis BE, Douglas C, Bohlmann J, Ritland K (2004) Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor Appl Genet 109:1283–1294
Santos AMM, Cavalcanti DR, Silva JMC, Tabarelli M (2007) Biogeographical relationships among tropical forests in north-eastern Brazil. J Biogeogr 34:437–446
Silva FAB (2011) First record of Coprophanaeus bellicosus (Olivier) (Coleoptera, Scarabaeidae) in a “Brejo de Altitude” forest in northeastern Brazil: a historical biogeographical approach. Rev Bras Entomol 55:615–617
Siqueira-Filho JA (2002) Bromélias em Pernambuco: Diversidade e aspectos conservacionistas. In: Tabarelli M, Silva JMC (eds) Diagnóstico da biodiversidade de Pernambuco. Massangana, Recife, pp 219–228
Su Y, Wang T, Deng F (2010) Contrasting genetic variation and differentiation on Hainan Island and the Chinese mainland populations of Dacrycarpus imbricatus (Podocarpaceae). Biochem Syst Ecol 38:576–584
Tabarelli M, Santos AMM (2004) Uma breve descrição sobre a história natural dos brejos nordestinos. In: Porto KC, Cabral JJP, Tabarelli M (eds) Brejos de altitude em Pernambuco e Paraíba: História Natural, Ecologia e Conservação. Brasília. Ministério do Meio Ambiente, p 324
Thomas E, van Zonneveld M, Loo J, Hodgkin T, Galluzzi G, van Etten J (2012) Present spatial diversity patterns of Theobroma cacao L. in the neotropics reflect genetic differentiation in pleistocene refugia followed by human-influenced dispersal. PLoS ONE 7:e47676
Vanzolini P, Williams EE (1970) South American anoles: geographic differentiation and evolution of the Anolis crysolepis species group (Sauria, Iguanidae). Arq Zool São Paulo 19:1–298
Wang X, Auler AS, Edwards RL, Cheng H, Cristalli PS, Smart PL, Richards DA, Shen C (2004) Wet periods in Northeastern Brazil over the past 210 kyr linked to distant climate anomalies. Nature 432:740–743
Whitmore TC, Prance G (1987) Biogeography and quaternary history in Tropical America. Oxford Science Publications/Clarendon Press, Oxford
Youens-Clark K, Buckler E, Casstevens T, Chen C, Declerck G, Derwent P, Dharmawardhana P, Jaiswal P, Kersey P, Karthikeyan AS, Lu J, McCouch SR, Ren L, Spooner W, Stein JC, Thomason J, Wei S, Ware D (2011) Gramene database in 2010: updates and extensions. Nucleic Acids Res 39(Database issue):D1085–D1094
Zellinger B, Akimcheva S, Puizina J, Schirato M, Riha K (2007) Ku suppresses formation of telomeric circles and alternative telomere lengthening in Arabidopsis. Mol Cell 27:163–169
Acknowledgments
The authors would like to thank the Laboratório de Análise Genética e Molecular of the Universidade Estadual de Campinas (UNICAMP) for support and infrastructure during the development of the SSR markers; to the MSc. Luiz Wilson Lima-Verde for his valuable help during the field work; the CNPq (National Council for Scientific and Technological Development) for the fellowship to LD; and PROPESQ/UFPE for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Liliane G. Dantas and Tiago Esposito have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dantas, L.G., Esposito, T., de Sousa, A.C.B. et al. Low genetic diversity and high differentiation among relict populations of the neotropical gymnosperm Podocarpus sellowii (Klotz.) in the Atlantic Forest. Genetica 143, 21–30 (2015). https://doi.org/10.1007/s10709-014-9809-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-014-9809-y