
Solid–state dissolution in N–methylmorpholine N–oxide was used to obtain solutions of mixtures derived from cellulose and various organosilicon additives, namely, tetraethoxysilane, vinyltriethoxysilane, and bis (trimethylsilyl)acetylene. Optical study of the phase composition and morphology of these solutions showed that they are two–phase emulsions with a rather broad size distribution of particles of the dispersed phase. The nature of the flow of the mixed systems in continuous and dynamic deformation when the rheological behavior is monotypic depends to a certain extent on the nature of the organosilicon additive. Dry wet–jet spinning was used to obtain composite fibers. The structure and morphology of these fibers were studied as well as their mechanical and thermal properties. Analysis of the x–ray patterns diffractograms of the cellulose and composite fibers showed that the introduction of organosilicon additives into the cellulose matrix leads to less structural ordering of the cellulose. The mechanical characteristics of the composite fibers show some decrease in the strength and deformation characteristics with an increase in the elastic modulus in comparison with the cellulose fibers. Heat treatment of the cellulose and composite fibers up to 1000°C revealed a significant increase in the mass of carbon residue, whose amount depends on the type of additive.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Lyocell fibers obtained from solutions in N–methylmorpholine N–oxide (NMMO) are considered potential precursors of carbon fibers and as an alternative to fibers obtained by the viscose method [1]. As in the case of viscose precursors, heat treatment of lyocell fibers is carried out using impregnation with pyrolysis catalysts [2]. Lewis acids, zinc chloride, calcium chloride, and ammonium phosphate have been proposed as such catalysts [3]. Silicon compounds occupy a special place among pyrolysis catalysts [4,5,6,7].
An alternative approach for the use of organosilicon additives involves the introduction of such compounds into the cellulose matrix during preparation of the spinning solutions [8,9,10]. Thus, the use of tetraethoxysilane permits not only control of the structure and mechanical properties of the cellulose precursors but also an increase in the carbon residue of the fibers upon their heat treatment. Unfortunately, there are no articles in the literature comparing the effect of the type of organosilicon additive on the rheological behavior, structure, thermal, and mechanical properties of fibers. In order to fill this gap, we studied the effect of alkoxysilanes, namely, vinyltriethoxysilane (VTEOS) and tetraethoxysilane (TEOS), which contain reactive and hydrolytically unstable Si–O–C bonds, and a chemically stable additive, bis (trimethylsilyl) acetylene (BTMSA), which lacks such bonds, on the process yielding precursor fibers and their properties.
Mixed solutions containing 18% cellulose (Baikal Pulp and Paper Mill) containing ~600 monomer units in the polymer, ~8% equilibrium moisture content, not less than 94% 〈–cellulose (GOST State Standard 6840–78), and organosilicon additives in amounts 5 to 30% of the cellulose mass in NMMO were prepared according to a solid–phase procedure [10, 11]. The NMMO sample from Demochem (China) contained 8–10% water and had a melting 44point of about 120°C. The organosilicon additives were obtained from Sigma–Aldrich (USA). The characteristics of these compounds are given in Table 1.
A system containing cellulose and an organosilicon additive was prepared prior to the solid–phase activation step. For this purpose, the additive was dissolved in acetone and mixed with powdered cellulose. Acetone was removed from the ternary system with rapid stirring and heating to yield a cellulose powder with the organosilicon additive. Then, this system was mixed with NMMO powder and subjected to mechanochemical activation under shear and pressure conditions. The resultant solid solution was melted at ~120°C and used as the spinning solution.
The extent of dissolution of cellulose in NMMO, phase composition, and morphology of the mixed solutions were studied using a Boetius polarization microscope (manufactured by Nadema in the former German Democratic Republic). Inhibition of thermooxidative decomposition was achieved by introducing 0.5% propyl gallate from Sigma–Aldrich.
The rheological properties of the cellulose solutions were studied using a Thermo Haake RheoStress rheometer (manufactured in Germany). For the measurements, we used a cone plane unit with 20 mm diameter (the angle between the cone former and plane was 1°). The testing was carried out under conditions of steady flow and a controlled shear rate. The rate range was from 0.001 to 100 sec–1. The testing temperature range was from 90 to 135°C. The dynamic testing was carried out in a regime corresponding to the range of linear viscoelasticity in the frequency region from 0.6 to 600 sec–1 at constant pressure 10 Pa.
Treatment of the homogeneous cellulose solutions with organosilicon additives was carried out using a CEAST Rheoscope 1000 capillary direct visualizing viscometer (manufactured in Italy). Formation of the cellulose and composite precursors (filler with hole diameter d = 1 mm (l/d = 40)) was carried out on the same instrument by wet dry–jet spinning. Drawing and winding of the fiber was carried out using the regular receiving shaft of the viscometer. The air gap between the capillary exit and the mirror of the deposition bath was 15 cm. The volume of the aqueous deposition bath was 180 ml. In order to exclude the accumulation of solvent in the deposition bath, we carried out the inlet of distilled water into the bath and the outlet of the aqueous solution of NMMO from the bath at a rate of about 2 ml/min.
The fiber take–up was 170 m/min. Water at ~22°C was used as the precipitating agent in the baths. Washing of the freshly formed fiber in water was carried out over 48 h. The fiber was dried at room temperature in the free state.
We studied cellulose fiber (CF) without additives and samples obtained in mixed solutions with 95/5 and 90/10 cellulose/additive component mass ratios (Table 2).
The morphology of the surface and cross–sections of the fiber was studied on a JEOL JSM U–3 scanning electron microscope (manufactured in Japan). The fiber structure was studied on a Rigaku microfocus copper rotating12–W anode x–ray generator (manufactured in Japan) in transmission mode at room temperature using CuKα radiation with wavelength λ = 1.542 nm. Parallel fiber fragment bundles (~100 pieces) were used to obtain diffractograms of the fibers.
The mechanical properties of the fibers were determined on an Instron 1122 tensile tester with 10 mm/min extension velocity according to GOST State Standard 10213.4.
The thermal behavior of the fibers was studied on a Mettler Toledo instrument with combined TGA/DSCI thermal analysis (manufactured in Switzerland). The measurements were carried out at from 30 to 1000°C at a heating rate of 10 deg/min. The argon flow rate was 10 cm3/min. A 70–μl aluminum oxide crucible was used.
Optical experiments showed that all the filled cellulose solutions with organosilicon additives are dimensionally biphasic throughout the entire concentration range studied. Thus, the particle diameter for the solution with 5 mass % VTEOS did not exceed 1 μm. Increasing the content of additive in the solutions led to a greater amount of these particles and an increase in their diameter up to 3 μm.
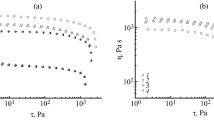
The heterophasic structure should affect the rheological properties of the mixed systems. Figure 1 shows curves for the dependence of the viscosity on the shear stress for the cellulose solutions with 5 and 10 mass % organosilicon additives.

Dependence of the shear viscosity on the shear pressure of the cellulose solutions with 5 (a) and 10 mass % organosilicon additive (b): 1) starting cellulose solutions, 2, 3, 4) cellulose solution with additives TEOS, BTMSA, and VTEOS, respectively, at 120°C.
Newtonian flow is seen at low shear velocities and stresses of the systems studied. The viscosity steadily decreases with an increase in the shear stress above 100 Pa.
The introduction of low–viscosity organosilicon additives to cellulose solutions in amounts not exceeding 6 mass % had virtually no effect on the flow characteristics of these solutions. A tendency toward the appearance of a nonlinear concentration dependence of the viscosity for TEOS and VTEOS with minima was noted when the contents of these compounds is 5.4 and 2.7 mass %, respectively. This behavior may be related to hydrolysis of the alkoxysilanes during dissolution and the formation of new compounds. In comparison with the alkoxysilanes, the silicon derivative of acetylene is resistant to the action of water and NMMO as indicated by the invariance of the viscosity upon change in the concentration of VTMSA (Fig. 2).

Concentration dependence of the viscosity of cellulose solutions with additives of TEOS (1), BTMSA (2), and VTEOS (3) at 120°C, σ = 20 Pa.
Since the viscosity in the presence of additives does not undergo major change, the formation of the fibers was carried out for all the systems under identical conditions.
Frequency dependence curves of the dynamic modulus at 120°C were obtained for all the solutions studied. As an example, the curves for solutions of cellulose with the TEOS additive are given in Fig. 3.

Frequency dependence of the dynamic moduli of starting cellulose solution (1) and solutions of cellulose with TEOS additive in amounts 5 (2), 10 (3), 15 (4), 20 (5), and 30 mass % (6) at 120°C.
For all the samples in the low frequency region, we find the inequality G” > G’, where G” is the mechanical loss modulus and G’ is the accumulation modulus, i.e., these solutions show predominantly viscous properties. The elastic component begins to exceed the viscous component when the testing frequency is increased to 50–100 sec–1. Such behavior is characteristic for flexible–chain polymers. The dynamic moduli of the solutions of cellulose with TEOS differ slightly from the corresponding values of the cellulose solution without additives. However, the nature of the curves for the solutions with and without additives is similar.
At low frequencies, the frequency dependence of the moduli is a power function with exponent 1.1 for the accumulation modulus and 0.6 for the loss modulus. These values are much lower than the traditional values of 2.0 and 1.0, which may indicate strong structure formation in the cellulose solutions both with and without additives. This behavior may be related to the presence of hydrogen bonds in the system, which are characteristic for starting cellulose. Although most of the hydrogen bonds are eliminated upon the dissolution of cellulose in NMMO, some of these bonds may be retained, leading to structure formation in the solution. Such structure formation is characteristic mainly under conditions of low–amplitude vibrations.
Curves for the temperature dependence of the viscosity in the range from 90 to 120°C were obtained for the cellulose solutions containing 5 mass % additive. These curves in coordinates of the Arrhenius equation [12] are shown in Fig. 4. These data were used to calculate the activation energy for viscous flow Ea. The values for Ea for all the solutions of cellulose with organosilicon additives were found to be lower than for solutions of cellulose without additive (Table 3). Thus, the introduction of slight amounts of low–molecular–weight components into the cellulose solution leads to lower values of Ea. The values of Ea for the composite samples vary slightly in the range from 68 to 76 kJ/mole, indicating an identical flow mechanism for these samples.

Dependence of the viscosity of the solutions of cellulose without additive (1) and with 5 mass % TEOS (2), BTMSA (3), and VTEOS (4) on inverse temperature.
The morphology of the composite fibers obtained by the wet dry–jet spinning method studied by scanning electron microscopy did not reveal major differences from CF. Microphotographs of the composite B–10 fibers are given as an example in Fig. 5. These microphotographs show that the cross–section of the fibers is round. There are no pronounced defects on the lateral surface. The mean fiber diameter ranges from 11 to 15 μm.

Microphotographs of composite B–10 fibers: a) cross–section and b) surface.
Structure is known to be one of the factors determining the mechanical and thermal properties of precursor fibers [13]. Less ordered structure is formed in the composite precursors than in CF as indicated by the change in intensity of the major peaks in the diffractograms as a result of disordering of the fibers (Fig. 6). Two major peaks corresponding to the cellulose II structure are seen in all the fiber diffractograms [14],

Equatorial diffractograms of cellulose (1) and composite fibers T–5 (2a), T–10 (3a), VT–5 (3b), B–5 (2c), and B–10 (3c).
The structural changes observed upon the introduction of organosilicon additives to a cellulose matrix are also reflected in the mechanical properties of the composite fibers (Table 4).
The structural data would lead us to expect some deterioration of the mechanical characteristics for the composite fibers. Indeed, the strength indices are most significantly lower for the VT fibers, while the introduction of 5 mass % BTMSA leads to only a slight decrease in strength. The strength indices for the fibers with TEOS are virtually the same as obtained for CF. The elasticity modulus of the fibers with additives TEOS and BTMSA are enhanced by 30–40%, while this value is on the level of CF for the most reactive VTEOS additive. A decrease of 20–30% for relative extension is observed for all the additives, which indicates the possible cross–linking of the cellulose molecules by the organosilicon additives.
The presence of additives in cellulose fibers affects their thermal behavior. As seen in Fig. 7, the carbon residue of the composite fibers is more than doubled. The introduction of organosilicon compounds into the bulk of cellulose fiber significantly shortens the combustion of cellulose and leads to an increase in the carbon residue by a factor of 2–2.5 [15].

Relative residual carbon mass content of cellulose (1) and composite fibers with additives of 5 mass % TEOS (2), VTEOS (3), and BTMSA (4) in a stream of 10 cm3/min argon with linear heating at a rate of 10 deg/min up to 1000°C.
Thus, the introduction of silicon compounds into cellulose does not lead to significant changes in the rheological behavior of the solutions in NMMO and, consequently, there is no need for special conditions for fiber fabrication. The structural organization of the composite fibers, strength and relative extension indices decrease with increasing additive content in the precursor but nevertheless remain high, which permits their conversion into carbon fibers. The chemical nature of the silicon–containing additives and their role as pyrolysis catalysts are most clearly seen in the considerable increase in the carbon residue obtained by heat treatment of the composite fibers up to 1000°C.
References
S. Peng, H. Shao, and X. Hu, J. Appl. Polymer Sci., 90, No. 7, 1941–1947 (2003), doi: https://doi.org/10.1002/app.12879.
G. Goldhalm, Lenzinger Ber., 90, 58–63 (2012).
A. G. Dumanle and A. H. Windle, J. Mater. Sci., 47, No. 10, 4236–4250 (2012), doi: https://doi.org/10.1007/s10853–011–6081–8.
M. E. Kazakov, A. M. Trushnikov, and M. L. Yunitskaya, Russian Federation Pat. 2045472 (1992).
A. M. Trushnikov, M. E. Kazakov, et al., Russian Federation Pat. 2047674 (1993).
P. Olri, Russian Federation Pat. 2256013 (2005).
J. Y. Hyuk, J. Y. Min, and L. S. Hong, J. Korean Soc. Safety, 31, No., 66–73 (2016), doi: https://doi.org/10.14346/JKOSOS.2016.31.066.
A. A. Konkin, Carbon and Other Heat–Resistant Fiber Materials [in Russian], Khimiya, Moscow (1974), p. 376.
I. Wizon and J. A. Robertson, J. Polymer Sci., Part C, No. 19, 267–281 (1967).
I. S. Makarov, L. K. Golova, et al., Fibre Chemistry, 49, No. 2, 101–107 (2017), doi: 10.1007/s10692–017–9851–5.
L. K. Golova, Russian Federation Pat. 1645308 (1992).
G. V. Vinogradov and A. Ya. Malkin, Polymer Rheology [in Russian], Khimiya, Moscow (1977).
V. G. Kulichikhin, I. Yu. Skvortsov, et al., Adv. Polymer Technol., 37, No. 4, 1099–1113 (2018), doi: https://doi.org/10.1002/adv.21761.
E. N. J. Ford and Sh. K. Mendon, et al., J. Eng. Fiber Fabr., 5, No. 1, 10–20 (2010).
I. S. Makarov, L. K. Golova, et al., Fibre Chemistry, 49, No. 4, 231-236 (2017), doi: https://doi.org/10.1007/s10692–018–9874–6.
This work was carried out with the financial support of the Russian Basic Research Fund, Grant No. 16–33–60218 mol-_a_dk.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimicheskie Volokna, Vol. 51, No. 1, pp. 26–30, January–February, 2019.
Rights and permissions
About this article

Cite this article
Makarov, I.S., Golova, L.K., Kuznetsova, L.K. et al. Composite Fibers From Cellulose Solutions with Additives of Bis (Trimethylsilyl) Acetylene and Alkoxysilanes: Rheology, Structure and Properties. Fibre Chem 51, 26–31 (2019). https://doi.org/10.1007/s10692-019-10041-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10692-019-10041-4