
A method for producing cellulose-based composite fibers with the addition of tetraethoxysilane (TEOS) with a high degree of dispersion of the additive in the cellulose matrix through a “solid phase” dissolution step of the cellulose in N-methylmorpholine-N-oxide (NMMO) is proposed. Investigation of the phase state and morphological features of mixtures of cellulose solutions with TEOS additives in NMMO has shown that in the entire range of concentrations studied, the solutions are biphasic emulsions, and their dispersity and the shape of dispersed phase droplets depend on the prehistory of solution preparation. It is established that the rheological behavior of cellulose solutions with TEOS additives varies depending on the amount of the additive. A theory about a mechanism for changing the rheological behavior of cellulose solutions with TEOS is formulated. X-ray diffraction analysis, scanning and transmission microscopy and IR spectroscopy were used to characterize the structure and morphology of composite fibers. The mechanical properties of composite cellulose fibers with TEOS are investigated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The main criterion determining the development of modern technology is the production of materials with the required set of properties. One of the main approaches to addressing this issue is the production of composite materials, in which carbon fiber (CF) used as the reinforcing phase is obtained from polyacrylonitrile (PAN) and hydrated cellulose (HC) precursor fibers [1].
Carbon fibers based on PAN fibers are characterized by high specific strength and rigidity. CF obtained from HC fiber has an increased coefficient of thermal expansion at moderate strength values, which allows using them in combination with silicon carbide fibers for thermal insulation of objects with a high intensity of heat release [2].
The main industrial method for the production of HC fiber is the viscose process. The ecological danger of the viscose process initiated research with the goal of finding new ways of processing cellulose into fibers. As a result of such investigations, an environmentally friendly process for the direct dissolution of cellulose in N-methylmorpholine-N-oxide (NMMO) has been developed and is now commercialized [3]. Cellulose fibers formed from solutions in NMMO have received the general name “lyocell” according to the classification of the international committee BISFA [4].
For the production of carbon fibers, viscose cord and technical lyocell yarn are used [5, 6]. The basic requirements for precursor viscose fiber and the technological process of obtaining CF based on it can be considered already formulated. Current work is focused mainly on optimization of the technological process by selecting catalysts and temperature-time regimes of pyrolysis and graphitization. Industrial development of the process of obtaining CF from lyocell fibers is at the initial stage of development. Published data show that the requirements for the structure and properties of viscose and lyocell precursors are largely the same. Thus, in [7] the authors showed that high molecular weight lyocell fibers with a fine-crystalline unstressed structure can be used as precursors for CF.
Prior to high-temperature treatment of HC, it is recommended to treat the precursors with so-called pyrolysis catalysts. The use of catalysts makes it possible to suppress the thermal effects of the main reactions of cellulose decomposition and thereby increase the yield of carbon, shorten the duration of processing, and obtain CF with the required mechanical properties [8]. The most common catalysts for pyrolysis are phosphorus- and nitrogen-containing compounds, salts of transition metals, etc. [9].
In the work performed by Konkin much attention was devoted to the production of CF in the presence of silicon oxides [10]. Subsequently, this approach has been widely developed [11]. Methods for depositing silica particles using the colloidal deposition method (hydrolysis of tetraethoxysilane) onto the surface of cellulose fibers, aerogels, powder, etc. are discussed in [12,13,14]. The authors of the patents [15, 16] showed that the use of substances containing silicon as catalysts is the most promising approach. This is due, first of all, to the quality of the produced carbon fibers, the environmental friendliness of the process and the availability of raw materials.
The processes of applying flame retardants to fibers, while of great importance for fiber thermolysis processes, have a number of significant disadvantages, such as uneven distribution on the fiber surface, inability to introduce an active substance into the volume of the precursor matrix, low concentrations of flame retardant solutions used, etc.
It is quite obvious that the most effective and promising prethermolysis treatment method of precursor fibers is the introduction of a fire retardant additive directly into the cellulose matrix, but, due to the complexity of this process, very little work has been done on this topic. Moreover, the works on the introduction of silicon additives into the polymer matrix do not address this problem in terms of obtaining precursor fibers. Thus, the effect of addition of organosilicon ethers, including tetraethoxysilane (TEOS) in the concentration range of 0.1-0.4%, on the structure and properties of composites based on thermosetting binders was investigated [17].
An attempt was made [18] to introduce silica nanoparticles into a cellulose matrix. An aqueous dispersion of silica with an average particle size of 7 nm was mixed with the system cellulose – 50% aqueous solution of NMMO. When excess water is removed, the mixture systems transition to viscous, filled cellulose solutions with SiO2 in NMMO. The introduction of silicon nanoadditives leads to disordering of the cellulose structure, which allows for a change of the mechanical properties of composite lyocell fibers and for regulating their fibrillation.
The objectives of this work are to develop a method for obtaining fiber precursors based on cellulose with the addition of TEOS through solid-phase cellulose solutions in NMMO, to investigate the phase state and rheological behavior of mixed solutions in NMMO, and to study the structure, morphology, and mechanical characteristics of the resulting composite fibers.
To prepare 18% mixed solutions of cellulose in NMMO, sulfate viscose powder cellulose from Baikal pulp and paper mill (DP = 700, H2O ~ 8%) was used as the matrix system, tetraethoxysilane with a molecular formula (C2H5O)4Si (“Sigma-Aldrich,” USA) was used as the composite additive, and N-methylmorpholine N-oxide (T m = 110-120 °C, H2O ~ 8-10%) (Demochem, China) was used as the direct cellulose solvent.
Solutions of Cellulose in NMMO were prepared by the previously developed method of “solid-phase” dissolution of cellulose in NMMO in the working assembly of a laboratory vibrating ball mill with preliminary activation of the NMMO – cellulose system under conditions of all-around compression and shear deformations [19]. To inhibit thermaloxidative degradation, 0.5% propyl gallate was added to the NMMO – cellulose system.
In the solid-phase activation process between cellulose and crystalline NMMO, the reaction of electron-donor-acceptor interaction proceeds to form H-complexes (solid solutions), which upon subsequent heating to 90-120 °C become viscous.
The complete dissolution of cellulose and the phase state of the mixture solutions were monitored with a Boetius polarization microscope (VEB Kombinat Nadema, GDR).
Fiber production was carried out on a dry-wet molding microstand made on the basis of an MV-3 capillary viscometer using single-channel spinnerets with different channel geometries (d = 200-1000 microns, l/d = 5).
The resulting highly homogeneous solutions were molded into an aqueous precipitation bath, where as a result of mass transfer, solvent was removed from the solution and coagulation of cellulose took place.
The rheological properties of cellulose solutions and filled solutions were studied using a rotational cone-plate rheometer “RheoStress600” (“ThermoHaake,” Germany) (working assembly diameter 20 mm, 1° angle between the cone generatrix and the plane). The tests were carried out under steady-state conditions in a controlled shear rate mode at a speed γ of 10–4 to 103 s–1 and a temperature of 120 °C.
The structure of the obtained fibers was studied by X-ray diffraction analysis (XRD). X-ray diffraction studies of fibers were carried out at room temperature in “transmission” mode on an X-ray DRON-3M instrument with CuKα radiation (λ = 1.542 Å). To obtain equatorial diffractograms of the fibers, parallel bundles of their fragments (50-60) were used.
IR spectra of cellulose fibers and composite fibers were collected by attenuated total reflection (ATR) using an infrared microscope “Hyperion-2000” coupled with an IFS-66 v/s Bruker FTIR spectrometer (100 scans, ZnSe crystal, 2 cm–1 resolution, 600-4000 cm–1 spectral range); instrument resolution was 0.5-2.0%.
Photomicrographs of the composite fibers were obtained using a “LEO 912AB Omega” transmission electron microscope (Carl Zeiss, Germany) with a LaB-based cathode (accelerating voltage 100 kV) and a “JSM U-3” scanning electron microscope (JEOL, Japan).
The mechanical characteristics of the fibers were determined using an “Instron 1122” tensile testing machine at a tensile strain of 10 mm/min in accordance with GOST 10213.4 – 2002, 10213.0 – 2002, 10213.2 – 2002.
To produce composite fibers based on cellulose with a silicon additive, three methods of introducing TEOS into the system were developed. The first method was based on the colloidal precipitation of TEOS particles on cellulose. To do this, TEOS was mixed with acetone, and then powdered cellulose was added to the resulting system. With vigorous stirring and heating, the acetone was removed from the ternary system, thus cellulose powder with a silicon additive was obtained. The second method involved the introduction of liquid TEOS into cellulose powder with vigorous stirring of the system. The mixture solutions were then prepared according to the procedure described in [20]. The third method consisted of adding TEOS to the viscous flowing cellulose solution in NMMO and mixing the resulting system.
The resulting filled solutions were biphasic in the entire investigated concentration range. Fig. 1 shows photographs of cellulose solutions in NMMO with 10% TEOS prepared according to the different methods.

Micrographs of filled 18% cellulose solutions in NMMO with 10% TEOS prepared according to the first (a) and third (b) methods.
As can be seen from the presented micrographs, the method of colloidal deposition of TEOS on cellulose (the first method) makes it possible to obtain cellulose solutions in NMMO with a better distribution of the additive. Separate particles with an average size of 2-3 microns can be seen in solutions prepared by this method. In solutions obtained by mechanically mixing the cellulose solution in NMMO with TEOS (third method), the average particle size reaches 20 microns. Particles of the additive in the NMMO – cellulose solution do not aggregate when the system is deformed.
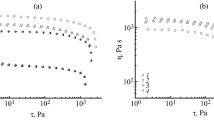
To assess the influence of TEOS on the behavior of cellulose solutions in NMMO during deformation, rheological properties of filled solutions were investigated on a rotational rheometer in the continuous shear deformation mode. As it turned out, the mixed solutions in NMMO with 5, 10 and 20% TEOS exhibit non-Newtonian behavior, i. e., their viscosity decreases with increasing shear stress. In this case, the nature of the flow curves does not change with an increase in the content of TEOS. As an illustration, Fig. 2 shows the flow curves of mixtures of cellulose and TEOS in NMMO. It can be seen that when TEOS is introduced into 18% cellulose solutions, the viscosity of the mixed solutions decreases, and if the content of the TEOS additive does not exceed 10%, the viscosity of the solutions does not change significantly, while when the content of TEOS is increased to 20%, the degree of viscosity drop significantly increases.

Flow curves for 18% cellulose mixtures with varying TEOS contents at 120 °C (in %): 1 – 0; 2 – 5; 3 – 10; 4 – 20.
The observed nature of the rheological behavior of mixtures is most likely due to the chemical transformations of TEOS, which take place in solutions of cellulose in NMMO at 120 °C. Organosilicon compounds in the presence of water are highly unstable, prone to hydrolysis reactions and subsequent condensation and polycondensation, forming oligomers and polymers with siloxane bonds of linear or cyclic structure [17, 21, 22]. In the presence of small amounts of water in an alkaline medium, the rates of hydrolysis and condensation of organosilicon compounds are low, with the formation of predominantly low molecular weight linear polymers and a small number of cyclic compounds.
In mixtures of cellulose and TEOS in NMMO, water, present in an amount of no more than 10-13%, is introduced into the system both by the cellulose itself and the hydrated form of the NMMO used. It is fair to assume that the presence of water, a basic solvent, and high temperature contribute to the conversion of TEOS into siloxane compounds. In the case of cellulose solutions with a low TEOS content not exceeding 10%, the phase of the resulting polysiloxanes is likely to be discrete, so its contribution to the flow of the system is negligible. With an increase in the TEOS content of up to 20% and, correspondingly, an increase in the fraction of the low viscosity phase of polysiloxanes, the probability of its flow in unique channels between the domains of the cellulose solution increases, which leads to a significant decrease in the viscosity of the system.
During molding mixed solutions into an aqueous precipitation bath, it appears that complete hydrolysis of TEOS and bonding of all the hydroxyl groups to form chains of polysiloxanes with side groups attached to silicon atoms occur simultaneously with coagulation of cellulose.
Transmission electron microscopy was used to estimate the shape, size and distribution of particles of the resulting polysiloxanes in the cellulose matrix (Fig. 3). As can be seen from the photomicrographs, in the fiber, in general, there are particles of polysiloxanes with an average size of 20-40 nm. A small number of the agglomerates observed in the field of view (Fig. 4) do not exceed 100-200 nm in size. Thus, the method of colloidal deposition of particles made it possible to obtain composite fibers with a high degree of dispersity of the filler particles.

A micrograph of a composite fiber consisting of 95% cellulose and 5% TEOS.

A micrograph of a composite fiber obtained from an 18% solution of cellulose in NMMO with TEOS (5% by weight of cellulose) in a light (a) and dark (b) field and diffraction of electrons by filler particles (c).
The structure of the filler particles was studied using an electron microscopy dark field method. It can be seen from Fig. 4 (a, b) that while only a clear shape of the particle is observed in a light field, in the dark field a scattering crystalline region is clearly manifested in this particle. This may indicate that the organosilicon filler particles formed in the cellulose matrix have an amorphous-crystalline structure. The electron diffraction pattern from the crystalline region of the particle (Fig. 4c) corresponds to the diffraction pattern of electrons on silicon oxide given in [23].
Using SEM, the morphology of composite fibers formed from an 18% solution of cellulose in NMMO with TEOS (5% by weight of cellulose) (T-5) was investigated. Fig. 5 presents comparative micrographs of composite and cellulose fibers. As can be seen, the surface of the fibers is smooth and even. The study of cross sections of composite and cellulose fibers revealed a circular cross-sectional geometry for all fibers under study.

SEM microphotographs of the surface and cross-sections of cellulose (a, b) and composite fibers T-5 (c, d).
Diffractograms of composite and cellulose fibers obtained by XRD analysis are shown in Fig. 6. The arrangement in the diffraction patterns of the main reflections in the region of 2θ ~ 12° (101), 2θ = ~ 20° (101) and 2θ ~ 21.5° (002) indicates that the composite and cellulose fibers have monotype structure characteristic of cellulose II [24]. The somewhat different ratio of the intensities of the equatorial peaks (101) and (002) of cellulose and composite fibers is probably due to a change in the intralayer ordering and a disruption in order along the axis of formation of the composite fiber.

Equatorial diffraction patterns of cellulose (a) and composite (b) T-5 fibers.
IR spectroscopy investigations of the structure of cellulose and composite fibers with the addition of 5% TEOS, same as XRD analysis, did not reveal any fundamental differences in the structure of these samples. The introduction of 15% TEOS to cellulose leads to a change in the intense bands characteristic of TEOS, and to the transformation of cellulose structure.
Fig. 7 shows the comparative IR spectra of cellulose and composite fibers obtained from 18% solutions of cellulose in NMMO with TEOS (15% of the weight of cellulose) and TEOS. Judging by the spectra of cellulose and composite fibers, their structural organization is markedly different. Thus, the strongest band in the TEOS spectrum attributed to Si–O–C bonds (1080-1100 cm–1) is present in spectrum 2, but it is shifted to the long-wavelength region up to 1043 cm–1 and overlaps with the strongest cellulose band at 1027 cm–1. This shift of the band is due to the transition from the Si–O–C bonds to the Si–O–Si bonds [25], which points to chemical transformations of TEOS into polysiloxanes, which take place in the processes of cellulose dissolution in NMMO and fiberizing. The presence in the spectrum 2 of bands from the valence (2931, 2974 cm–1) and deformation (1390 cm–1) vibrations of alkyl groups, as well as the 797 cm–1 band absent in the cellulose spectrum, but clearly visible in the spectrum of a composite fiber, apparently, is due to the preservation of alkyl groups of the original TEOS as side substituents of silicon atoms in the formed silsesquioxane chains.

IR spectra of cellulose fibers (1), composite cellulose fibers with 15% TEOS (2) and TEOS (a drop between KBr plates) (3).
Despite the hydrophobic nature of silsesquioxanes, their presence in the cellulose matrix results in a change in the structure of the cellulose. To estimate the degree of ordering of cellulose polymer chains, the bands at 2900 cm–1 (νCH) and 1370 cm–1 (corresponding to deformational vibration of a complex shape involving the deformation of –OH, –CH and –CH2 groups) are often used as internal standard bands. The relative intensity of the 1370/2900 cm–1 bands is the most sensitive measure of the degree of crystallinity of cellulose. The 1430 cm–1 band belongs mainly to the deformational vibrations of the CH2 group, and the 900 cm–1 band is responsible for the amorphous sections of cellulose, so this parameter corresponds more to lateral ordering.
It can be seen from the spectra shown in Fig. 7 that when 15% TEOS is added to the cellulose, characteristic changes occur in the characteristic cellulose absorption bands. The greatest changes were noted in the 1200-1450 cm–1 region sensitive to the crystalline phase of cellulose. Judging by the spectra, the degree of crystallinity upon the introduction of TEOS decreases. At the same time, the intensity of the bands characteristic of the amorphous sections of the cellulose chain increases: 1152, 900 cm–1. The same patterns are demonstrated by the data presented in Table 1. The crystallinity index of the system shown in Table 1 was determined from the relative intensities of the bands 1370/2900 (second line) and the O’Connor crystallinity index (first line) determined from the relative intensities of the 1430/900 bands and the intensity of the bands characterizing the amorphous phase (1152 and 900 cm–1). Thus, the introduction of 15% TEOS into cellulose results in a loss of crystallinity and an increase in the amorphous phase in the cellulose matrix.
The mechanical characteristics of cellulose and composite fibers are shown in Table 2. As can be seen from Table 2, the mechanical parameters of composite fibers completely correspond to the results of structural studies, namely: introduction of up to 5-10% TEOS into the cellulose matrix slightly affects the strength and modulus of elasticity, while the deformation properties of composite fibers increase threefold. An increase in the content of the additive to 15% leads to an ~ 8% decrease in strength and an almost two-fold decrease in the modulus of elasticity. In this case, the elongation at break increases to 37%. Thus, compounds of the siloxane series formed as a result of chemical transformations of TEOS in the processes of dissolution and coagulation of cellulose upon contact with an aqueous precipitation bath play the role of plasticizing additives [18].
Based on the obtained data, it can be concluded that for the production of precursors of carbon fibers based on cellulose and TEOS, the concentration of the organosilicon TEOS in the cellulose matrix should not exceed 15%.
References
A. I. Kadantseva and V. A. Tverskoi, Carbon Fibers [in Russian], textbook, Moscow State University of Fine Chemical Technologies named after M.V. Lomonosov, Moscow (2008) 55 p.
A. V. Skolunov and M. E. Kazakov, Fibre Chemistry, 32, No. 5, 365-371 (2000).
L. K. Golova, V. G. Kulichikhin, S. P. Papkov, Vysokomol. Soed. Ser. A., 28, No. 9, 1795-1809 (1986).
Proceedings of 92nd Meeting of the BISFA Subcommittee Terminology, Paris, France, 1989.
S. Peng, H. Shao, and X. Hu, J. Appl. Polymer Sci., 90, No. 7, 1941-1947 (2003).
V. G. Kulichikhin, L. K. Golova, et al., Polymer Sci. Ser. C., 58, No. 1, 74-84 (2016).
Yu. V. Karasev et al., Russian Federation Patent 2429316 C1 (2010).
V. V. Korshak, Chemical Structure and Temperature Characteristics of Polymers [in Russian], Nauka, Moscow (1970) 420 p.
D. N. Chernenko, dissertation, May 16, 2006, NIIgrafit, Moscow (2015) 240 p.
A. A. Konkin, Carbon and Other Heat-Resistant Fibrous Materials [in Russian], Khimiya, Moscow (1974) 376 p.
P. Olri, Russian Federation Patent, 2256013 (2005).
Zunli Mo, Zhongli Zhao, et al., Acta Mater. Compos. Sinica., 25, No. 4, 24-28 (2008).
Arnaud Demilecamps. Synthesis and characterization of polysaccharide-silica composite aerogels for thermal superinsulation. Materials. Ecole Nationale Superieure des Mines de Paris, 2015. English. <NNT : 2015ENMP0029>.
S. Sequeira, D. V. Evtuguin, and I. Portugal, Polymer Compos., 30, No. 9, 1275-1282 (2009).
M. E. Kazakov, A. M. Trushnikov, and M. L. Yunitskaya, Russian Federation Patent, 2045472, (1992).
A. M. Trushnikov, M. E. Kazakov, et al., Russian Federation Patent, 2047674, (1993).
I. Yu. Skvortsov, L. B. Kandyrin, et al., Vestnik MITKhT, 5, No. 4, 98-100 (2010).
P. Kulpinski, J. Appl. Polymer Sci., 98, 1793-1798 (2005).
L. K. Golova, Russian Federation Patent, 1645308 (1992).
L. Golova, I. Makarov, et al., Cellulose – Fundamental Aspects, InTech, Rijeka (2013) 303 p.
I. A. Karpov, E. N. Samarov, et al., Fiz. Tverd. Tela, 47, No. 2, 334-338 (2005).
R. Sato-Berru, J. M. Saniger, et al., J. Mater. Sci. Eng. A-Struct., 3, No. 4, 237-242 (2013).
J. Zhu, J. Wu, et al., J. Mater. Sci., 45, No. 24, 6769-6774 (2010).
E. N. Johnson Ford, Sh. K. Mendon, et al., J. Eng. Fiber Fabr., 5, No. 1, 10-20 (2010).
L. Bellamy, Infrared Spectra of Complex Molecules [in Russian], Izdatinlit, Moscow (1963) 590 p.
The authors express their gratitude to B. F. Shklyaruk for conducting X-ray diffraction analysis of composite fibers. The work was supported by the Russian Foundation for Fundamental Research. Grant 16-33-60218 mol_a_dk.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimicheskie Volokna, No. 2, pp. 24 – 30, March – April, 2017.
Rights and permissions
About this article

Cite this article
Makarov, I.S., Golova, L.K., Kuznetsova, L.K. et al. Composite Fibers Based on Cellulose and Tetraetoxysilane: Preparation, Structure and Properties. Fibre Chem 49, 101–107 (2017). https://doi.org/10.1007/s10692-017-9851-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10692-017-9851-5