
Abstract
Linseed (Linum usitatissimum L.) is an important oilseed as well as stem fiber crop and rich source of omega-3 fatty acid. The present study aims to develop linkage map based on Indian genotypes and utilize it for mapping QTLs for important agronomic traits. Two diverse parental genotypes (KL-213 and RKY-14) of linseed showed wide range of variability for oil content and yield attributes. These parental genotypes also showed reasonable level of SSR polymorphism (~ 9.0%). The mapping population showed normal distribution of phenotypic traits. One hundred forty-six SSR markers were mapped on 15 linkage groups with marker density ranging from 3 to 18 markers per linkage group at average distance of 14.2 cM. A total of 11 QTLs were identified for six quantitative traits. Three QTLs for capsules/plant, 2 QTLs each for plant height, seeds/capsule and oil content and 1 QTL each for branches/plant and seed weight/plant were detected. Phenotypic variability explained by these QTLs varied from 1 to 15.23%. This study provides framework linkage map of linseed using Indian genotypes, which needs to be enriched further for future application in marker assisted breeding of linseed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Linseed (Linum usitatissimum L., 2n = 30), also referred as flax, is an annual, self-pollinated crop being utilized as both oilseed and fiber crop. Fiber and oilseed flax belongs to the same species but both are morphologically different. Linseed type is more branched and short heighted than fiber type (Gill 1987). Flax seed contain about 45–50% oil; significantly more than fiber varieties that contains 35–42% oil (Cloutier et al. 2011). Linseed oil consists of mainly five fatty acids i.e. palmitic acid (~ 6%), stearic acid (~ 4.4%), oleic acid (~ 24%), linoleic acid (~ 15%) and linolenic acid (~ 55%) (Westcott and Muir 2003). Linseed oil is utilized for drying oil, paints, linoleum flooring, inks, varnishes and soaps due to high proportion of linolenic acid (Cullis 2007). Currently, it is primarily grown in Canada which is the world’s largest producer of linseed (FAOSTAT 2014). Globally, in linseed production India ranks 6th, from an area of 0.284 million hectare (FAOSTAT 2014). There is huge gap in yield of linseed among top six linseed growing countries. The average yield of linseed in Canada is ~ 1.4 t/ha while in India it is only 0.49 t/ha (FAOSTAT 2014). The yield is one of the most important traits for genetic improvement. Not much effort has been made to improve yield potential of linseed (seed and oil) in India. This crop has huge potential for industrial applications as well as for human consumption. Therefore, there is need to explore the ways and impart new tools to improve the yield of the linseed. The conventional breeding methods with application of molecular tools could provide a better option for its faster and precise genetic improvement. Understanding the genetic diversity and genetic architecture of concerned crop plant is a fundamental approach for devising suitable breeding strategies towards its genetic improvement. Mapping QTLs for important agronomic traits is a potential tool to dissect the genetic architecture and laid down the basis of marker assisted selection (MAS) in crop breeding. In linseed, various molecular markers have been deployed over various set of genetic material to evaluate the level of genetic variation (Adugna et al. 2006; Fu 2006; Cloutier et al. 2009; Uysal et al. 2010; Deng et al. 2010, 2011; Soto-Cerda et al. 2011a, b; Pali et al. 2014, 2015; Chandrawati et al. 2014, 2017; Dash and Samal 2016). But limited studies are reported regarding QTL mapping. Till date only four genetic maps of linseed have been reported (Spielmeyer et al. 1998; Oh et al. 2000; Cloutier et al. 2011; Kumar et al. 2015). The first map has been developed using AFLP markers for Fusarium wilt resistance (Spielmeyer et al. 1998). Oh et al. (2000) used RFLP and RAPD for linkage mapping and identification of markers linked to loci controlling the heritable changes in response to environmental growth conditions. Cloutier et al. (2011) developed SSR based linkage map and identified QTLs for fatty acids. Recently, Kumar et al. (2015) constructed SSR and SNP based map to identify markers for fatty acids and yield related traits. However, none of these studies used any Indian genotypes. Therefore, it is pertinent to perform QTL mapping in linseed for important agronomic traits of Indian genotypes. The present investigation is carried to developed SSR based linkage map of linseed, using Indian genotypes and to detect QTLs for oil content and yield attributes.
Materials and methods
Parental lines and mapping population
The parental lines used in the present investigation include genotype RKY-14 (~ 44% oil content) and KL213 (~ 32% oil content) (Chandrawati et al. 2014). Apart from oil content, these parental lines also had contrasting traits such as days to flowering, plant height, seed weight/plant and capsule weight/plant. The crossing was done manually by hand pollination. The F1 hybrid plants were raised and selfed to obtain the seeds of F2 progeny. A total of 200 F2 plants were raised in field at CSIR-National Botanical Research (NBRI), Lucknow at a spacing of 15 cm within rows and 45 cm between rows. Out of 200, 140 plants of F2 population survived in the field. The leaves from these plants were collected at seedling stage. Data on morphological traits, namely days to flowering, plant height, branches/plant, capsules/plant, capsule weight/plant, seed weight/plant, number of seeds/capsule and test weight were recorded. The oil content was measured in percentage through Nuclear Magnetic Resonance (NMR) spectrometer (Bruker Corporation, USA).
DNA extraction and SSR genotyping
The genomic DNA was extracted from fresh and young leaves following CTAB method of Saghai-Maroof et al. (1984) with minor modifications. Briefly, 5 g leaves were grinded in liquid nitrogen along with 2% polyvinylpyrrolidone (PVP) and transferred in 15 ml of pre-warmed extraction buffer and incubated for 45 min at 60 °C in water-bath. Equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) was added and centrifuged for 20 min at 10,000 rpm at room temperature (25 °C). Two volume of chilled isopropanol was added to precipitate the DNA. The DNA pellet was washed with 70% ethanol, air dried and dissolved in 500 µl of sterile water. The extracted genomic DNA was treated with RNase A and incubated at 37 °C for 1 h. Equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) was added, centrifuged at 10,000 rpm for 5 min and upper aqueous layer was transferred in new tubes. Two volume of chilled absolute alcohol containing 1/10th volume of 3 M sodium acetate was added and centrifuged at 10,000 rpm for 5 min to precipitate the DNA. Finally, the DNA pellet was washed 2 times with 70% ethanol, air dried and dissolved in 500 µl sterile water. The concentration of genomic DNA was determined using a Nanodrop spectrophotometer ND1000 (Nanodrop Technologies, DE, USA) and quality was checked by running on 0.8% agarose gel. Finally, DNA was normalized to 10 ηg/µl for PCR amplification.
A total of 1720 SSRs were used for polymorphism screening between parental genotypes. Out of 1720 SSRs, 397 SSR primers were selected from previous reports (Cloutier et al. 2012b) based on distribution on linkage groups and rest 1323 SSRs extracted from whole genome sequence data. For SSR identification from genome sequence, the 88,420 scaffold sequences (~ 3.2 GB) were downloaded (www.phytozome.net) and subjected to SSR search using MISA (http://pgrc.ipk-gatersleben.de/misa) with search criteria of a minimum of eight repeat unit in case of dinucleotide, five in case of tri and tetranucleotide and four in case of penta and hexa-nucleotide SSRs. The primers were designed for SSR containing sequences using PRIMER3 (http://frodo.wi.mit.edu/primer3) with major criteria as: length, 20–26 bp; melting temperature, 55–65 °C and length of PCR product 100–400 bp. We followed the genotyping method of Schuelke (2000) and thus all the forward primers were synthesized with additional 18 base tag (5′ TGTAAAACGACGGCCAGT 3′) as M13 tail. In addition, same sequence of 18 base tag was also synthesized with four fluorescent dye i.e. FAM, VIC, NED and PET.
The PCR amplification was carried out in 10 µl reaction volume that contained 10 ηg of genomic DNA, 1× PCR master mix (AmpliTaq Gold®, Applied Biosystems, USA), 0.1 µl (5 pmol/µl) of forward primer (tailed with M13 tag), 0.3 µl (5 pmol/µl) each of both, reverse primer and M13 tag (labeled with either 6-FAM, VIC, NED and PET). PCR was performed on Veriti Thermal Cycler (Applied Biosystems, USA) using following condition: initial denaturation at 95 °C for 5 min followed by 36 cycle of 94°C for 30 s, 50–55 °C (primer specific) for 45 s and 72 °C for 1 min. Subsequently, 10 cycles of denaturation for 30 s at 94 °C annealing for 45 s at 53 °C, extension for 45 s at 72 °C followed by final extension for 15 min at 72 °C was performed. The PCR products were first checked for their amplification on 1.5% agarose gel, then post PCR pool was prepared based on fluorescence labeled primers. For post PCR pooling, 1 µl of 6-FAM and 2 µl of each VIC, NED and PET labeled PCR product with different SSRs were combined with 13 µl of water. Then 1 µl of that mix was added to 10 µl Hi-Di formamide containing 0.25 µl GeneScan™ 600 LIZ® as internal size standard. This was then denatured for 5 min at 95 °C, quick chilled on ice for 5 min and loaded on ABI 3730xl DNA Analyzer for capillary electrophoresis. The raw data was analyzed by GeneMapper v4.0 software (Applied Biosystems, Foster City, CA, USA) to obtain allele size.
Construction of framework linkage map
The genotypic data of SSR marker derived over 140 F2 plants were tested for Mendelian segregation. The raw data were subjected to test of significance using Chi square test for goodness of fit. The genotypic data showing Mendelian segregation were selected for linkage mapping. The linkage map was constructed with MAPMAKER v3.0 (Lander et al. 1987) using genotypic data of 140 F2 plants with 161 polymorphic SSRs (Supplementary Table 1). Mapping was done at maximum of 50% recombination and a LOD of 3.0, a marker position within a linkage group was determined with the ‘RIPPLE’ command and the ‘GROUP’ command was used for the best marker order of the linkage group. Map distance was calculated using Kosambi mapping function (Kosambi 1994) and centimorgan unit was used. Graphic representation of linkage groups was drawn using software MapChart version 2.2 (Voorrips 2006).
QTL mapping
Single locus QTL mapping was conducted through composite interval mapping (CIM) using QTL Cartographer version 2.5 (Wang et al. 2007) for nine traits. A stringent experimental LOD threshold > 2.5 for the detection of definite QTL were calculated based on 1000 permutation at P < 0.05. The relative contribution of a genetic component (R2) was calculated as proportion of the phenotypic variance explained (PVE). QTL explaining more than 10% phenotypic variation (PV) were considered as major QTL.
Results
Phenotypic variability
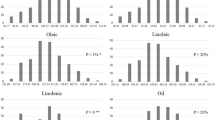
The two parental lines had wider range for oil content (KL-213: ~ 30% and RKY-14: ~ 44%) as well as for other important traits (Fig. 1, Table 1). The F2 plants derived from these genotypes also showed wide range of trait variability and had normal distribution of data for the nine traits evaluated, although transgressive segregation was also observed for each trait (Fig. 1). The minimum, maximum and mean value of different traits of mapping population is presented in Table 1. The value of skewness and kurtosis were generally less than 1.0, indicating suitability of data for QTL analysis.

Frequency distribution of the mean values of the morphological traits in the F2 mapping population derived from the cross KL-213 × RKY-14
Marker polymorphism and segregation analysis
Total 1720 SSR markers were screened for polymorphism between parental lines and of which, 161 SSR were found to be polymorphic showing 9.4% polymorphism. Among these polymorphic SSRs, 74 (4.30%) were from previously reported SSRs (Cloutier et al. 2012b) and 87 (5.05%) were novel SSRs. The genotypic data of 161 SSRs with 140 F2 plants subjected to Chi square test for goodness of fit. Fifteen SSRs showed significant distortion from Mendelian segregation and thus excluded in further analysis. Finally, 146 markers used for construction of framework linkage map of linseed and QTL identification.
Construction of framework linkage map
A total of 15 linkage groups (LG1 to LG15) were obtained at LOD > 3.0 (Fig. 2) harboring 146 SSRs. The linkage map spans a total genetic distance of 2074.2 cM, with an average locus spacing of 14.2 cM (Fig. 2). The 146 mapped markers included 69 previously reported SSRs (Cloutier et al. 2012b) and 77 novel SSRs. Each linkage group differed from each other with respect to total number of markers mapped, total cM distance and marker density. A large variation in length was exhibited by these fifteen linkage groups, which varied from a minimum of 19 cM (LG15) to a maximum length of 280 cM (LG1). The average marker density on each linkage group revealed that the markers were distributed randomly and unevenly. The marker density was highest in LG1 (18 markers) and lowest in LG15 (3 markers). The marker distribution on linkage groups revealed that 43 previously reported markers were mapped on the same linkage groups in the present study and 23 occupied different linkage group (Supplementary File 1).

Framework linkage map of linseed prepared using 140 F2 population derived from cross KL-213 × RKY-14 and 146 SSR markers. Genetic distance are indicated on left in cM and marker at right side. Different QTLs are depicted triangle and name
QTL identification for agronomic traits
QTL analysis for nine different traits namely: days to flowering, plant height, number of branch/plant, number of capsule/plant, capsule weight/plant, seed weight/plant, test weight, number of seeds/capsule and oil content was performed. A total of 11 QTLs were identified for six traits (Table 2). No QTL could be detected for days to flowering, capsule weight/plant and test weight. Two QTLs were identified for plant height (QPh.1 and QPh.2) at LOD of 3.58 and 3.06 respectively. The Qph.1 was identified on LG3 at position of 50.4 cM with confidence interval of 38.0–62.1 cM and closest to marker LUSc_149_01. The QPh.2 was detected on LG5 at position 20.4 cM with confidence interval of 14.0 to 44.0 cM and closest to marker LU_2497. The QTL QPh.2 explained 15.23% of the phenotypic variance with negative additive and positive dominance effect. Single QTL QBp.1 was detected for number of branches/plant on LG7 (LOD > 3.03) at position of 17.4 cM with confidence interval of 3.2–25.6 cM. with closest marker LU_3017. This QTL explained 4.64% of the phenotypic variance with positive additive and dominance effect. Three QTLs were identified for number of capsules/plant as QCp.1, QCp.2 and QCp.3. The QCp.1 was identified on LG2 position at 42 cM, with confidence interval of 36.8–68.8 cM at LOD value of 2.71. This QTL explained 6.92% of the phenotypic variance with negative additive and dominance effect. The QTL QCp.2 was detected on LG2 at position 141.6 cM, with confidence interval 105.6–155.2 cM at LOD value of 2.8 with closest marker LU_344. The QTL QCp.2 explained 6.27% of the phenotypic variance with positive additive and dominance effect. The QTL QCp.3 was detected on LG6 at position of 38 cM at LOD value of 2.61, with confidence interval of 22.4–54.4 cM. Single QTL QSw.1 was detected on LG 3 at a position of 145 cM at LOD value of 2.73 for seed weight/plant with confidence interval of 137.6–180.0 cM and 10.3% of the phenotypic variance with their positive additive and negative dominance effect. Two QTLs, QSpc.1 and QSpc.2 were detected for number of seeds/capsule at LOD > 2.52 and > 2.62, respectively. The QSpc.1 was identified on LG11 at position of 34 cM, with confidence interval of 18.0–42.0 cM. This QTL explained 11.1% of the phenotypic variance with positive additive and negative dominance effect. The QTL QSpc.2 was detected on LG13 at position of 50.1 cM with confidence interval of 46.0–64.1 cM and with 12.75% of the phenotypic variance with negative additive and positive dominance effect. Two QTLs, QOc.1 and QOc.2, were identified for oil content at LOD > 2.85 and 3.21 respectively. The QOc.1 was identified on the LG10 at position of 26.5 cM, with confidence interval of 10.0–42.0 cM. The QTL QOc.2 was detected on LG12 at 54.9 cM, with confidence interval of 44.6–72.1 cM. The QTL explained 8.39% of the phenotypic variance with negative additive and positive dominance effect.
Discussion
Most of the agronomically important traits are polygenic in nature and have complex inheritance which complicates the selection for genetic improvement. The marker assisted selection (MAS) is an alternative selection tool which allows the acceleration of the breeding program and substantial reduction of time, cost and space for plant development. The construction and development of genetic maps is important tool for the identification of quantitative trait loci (QTL) underlying the traits. To date only four genetic maps of linseed are developed and published. The first map based on RFLP and RAPD markers spanned 1000 cM with an average spacing of 10.6 cM and consist 15 linkage groups with 94 markers. The second linkage map covered 1400 cM with an average of 10 cM and consist 18 linkage groups with 213 AFLP markers. The other two maps were developed using SSR and SNP markers. The first microsatellite based linkage map of linseed consists of 113 EST-SSRs markers mapped to 24 linkage groups (Cloutier et al. 2011) spanning ~ 833.8 cM and used to detect QTLs for fatty acid traits. Two QTLs each for linoleic acid, linolenic acid, and iodine value and 1 QTL for palmitic acid was identified. An integrated consensus linkage map from three linseed mapping population was developed by Cloutier et al. (2012b) incorporating 770 markers based on 371 shared markers including 114 that were shared by all three populations and 257 shared between any two populations on the 15 linkage group. The length of map was 1551 cM with an average of 2.0 cM which correspond to ~ 74% of the linseed genome size of 370 Mb. Later, Kumar et al. (2015) developed linkage map using 329 SNP and 362 SSRs markers distributed across 15 linkage groups. They detected 20 QTLs corresponding to 14 traits. The present investigation is the first report of SSR-based linkage map utilizing Indian genotypes which has the potential to be useful further in linseed breeding. This SSR based map covers a total of 2074.2 cM distance (Fig. 2) and 15 groups with an average of 14.2 cM slightly higher length than reported previously (Spielmeyer et al. 1998; Oh et al. 2000; Cloutier et al. 2011; Kumar et al. 2015). Linseed is a true diploid plant with 15 chromosome pairs (Cullis 2007). The present map comprises 15 linkage groups indicating that the marker density is not yet sufficient and required the additional markers in order to increase the coverage of the genome and decrease spanning distance of the markers in this framework linkage map.
The characterization and polymorphism of SSRs helps in the identification of suitable markers, which will further be used in various genetic studies. In the present investigation, all the identified SSRs were subjected to PCR optimization, amplification and polymorphism detection and studies for various attributes. Total of 1720 SSRs identified from previous report and whole genome sequence of linseed from database and subjected for PCR optimization and polymorphism detection in L. usitatissimum. The polymorphism screening was carried out between genotypes KL213 and RKY-14, which were used as parental lines for developing mapping population segregating mainly for oil content and also for some other agronomic traits. A total of 15, out of 161, markers (9.31%) deviated significantly from expected Mendelian segregation of 1:2:1 in F2 plants. Segregation distortion has been observed in other crops also including maize (19–36%, Lu et al. 2002), Medicago truncatula (27%, Thoquet et al. 2002), common bean (37.3%, de Campos et al. 2011), peanut (8.5–22.8%, Hong et al. 2010), Brassica rapa (2.6%, Song et al. 1991), grapevine (7–11%, Doligez et al. 2006). Hackett and Broadfoot (2003) suggest that population size influence the segregation distortion when two markers are separated by more than 10 cM. The marker distortion percentage was found to be lower than that reported previously in linseed (Cloutier et al. 2011; Spielmeyer et al. 1998).
QTL mapping has been used as an efficient approach to analyze the quantitative traits in plants. To identify SSRs markers associated with important traits, the linkage map developed in the present study was used to detect QTLs for different morphological traits. In linseed, several molecular markers such as RAPD, RFLP, AFLP and SSR have been developed and used for various genetic studies (Spielmeyer et al. 1998; Oh et al. 2000; Cloutier et al. 2009, 2011, 2012a; Everaert et al. 2001; Fu et al. 2002a, b; Adugna et al. 2006; Roose-Amsaleg et al. 2006). However, till date only very limited QTL maps have been reported and none of them were utilized genotypes from India. There have been QTLs reported for resistances to Fusarium wilt (Spielmeyer et al. 1998) and linolenic acid (Cloutier et al. 2011). Recently, Kumar et al. (2015) identified 20 QTLs for 14 traits in 243 individual of RIL population. Here, we developed SSR based linkage map of linseed utilizing Indian genotypes and identified QTLs for important agronomic traits. A total of 11 QTLs (Table 2) were detected for 6 traits (plant height, number of branch/plant, number of capsule/plant, seed weight/plant, number of seed/capsule and oil content) which were sparsely distributed on the 9 linkage groups. Two QTL for number of capsule/plant were located on LG2 and 1 QTL for plant height and 1QTLfor seed weight/plant located on LG3. Cloutier et al. (2011) detected 2 QTLs each for linoleic acid and linolenic acid on LG7 and LG16. Soto-Cerda et al. (2014) detected 3 QTL for linoleic acid on LG3, LG5 and LG12. Out of 2 QTLs for plant height, QPh.2 on LG5 could be considered as major QTL as it explains 15.23% of the total phenotypic variation. None of the other QTLs explains such higher phenotypic variation. The 2 QTLs (QSpc.1 and QSpc.2) identified for number of seeds/capsule was also found to be important as they explain comparatively higher phenotypic variation. Similarly, one QTL for seed weight/plant (QSw.1) and one QTL of oil content (QOc.2) explains considerable phenotypic variation and thus could be potential QTLs for future studies and applications. Cloutier et al. (2011) reported 2 major QTL for linoleic and linolenic acid with 34 and 20% of the phenotypic variability. Most of the previous studies reported QTLs for fatty acid composition in linseed. Kumar et al. (2015) reported 1 QTL for oil content on LG8 accounting for 13% of phenotypic variation. However, we found only ~ 8% of phenotypic variation for oil content QTL. The low percentage of variability explained by QTLs associated with oil content might be due to the parental genotypes used in mapping population having not much difference in oil content and or low density of mapped markers on linkage group. To identify fine QTLs for oil content and other important traits, high density linkage map needs to be developed. This study presents a framework linkage map based on Indian linseed genotypes, which needs to be further enriched with more number of SSR/SNP markers in future and could be utilized for fine mapping of QTLs for marker assisted genetic improvement of linseed.
References
Adugna W, Labuschagne MT, Viljoen CD (2006) The use of morphological and AFLP markers in diversity analysis of linseed. Biodivers Conserv 15:3193–3205
Chandrawati, Maurya R, Singh PK, Ranade SA, Yadav HK (2014) Diversity analysis in Indian genotypes of linseed (Linum usitatissimum L.) using AFLP marker. Gene 549:171–178
Chandrawati, Singh N, Kumar R, Kumar S, Singh PK, Yadav VK, Ranade SA, Yadav HK (2017) Genetic diversity, population structure and association analysis in linseed (Linum usitatissimum L.). Physiol Mol Biol Plants 23:207–219
Cloutier S, Niu Z, Datla R, Duguid S (2009) Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor Appl Genet 119:53–63
Cloutier S, Ragupathy R, Zhixia N, Duguid S (2011) SSR-based linkage map of flax (Linum usitatissimum L.) and mapping of QTLs underlying fatty acid composition traits. Mol Breed 28:437–451
Cloutier S, Miranda E, Ward K, Radovanovic N, Reimer E, Walichnowski A, Datla R, Rowland G, Duguid S, Ragupathy R (2012a) Simple sequence repeat marker development from bacterial artificial chromosome end sequences and expressed sequence tags of flax (Linum usitatissimum L.). Theor Appl Genet 125:685–694
Cloutier S, Ragupathy R, Miranda E, Radovanovic N, Reimer E, Walichnowski A, Ward K, Rowland G, Duguid S, Banik M (2012b) Integrated consensus genetic and physical maps of flax (Linum usitatissimum L.). Theor Appl Genet 125:1783–1795
Cullis CA (2007) Flax. In: Kole C (ed) Genome mapping and molecular breeding in plants, vol 2. Springer, Berlin, pp 275–295
Dash M, Samal KC (2016) Agro-molecular diversity analysis of linseed (Linum usitatissimum L) genotypes using microsatellite based simple sequence repeats (SSR) marker. J Plant Sci Res 32:87
de Campos T, Oblessuc PR, Sforça DA, Cardoso JM, Baroni RM, de Sousa AC, Carbonell SA, Chioratto AF, Garcia AA, Rubiano LB, de Souza AP (2011) Inheritance of growth habit detected by genetic linkage analysis using microsatellites in the common bean (Phaseolus vulgaris L.). Mol Breed 27:549–560
Deng X, Long SH, He DF, Li X, Wang YF, Liu J, Chen XB (2010) Development and characterization of polymorphic microsatellite markers in Linum usitatissmum. J Plant Res 123:119–123
Deng X, Long SH, He DF, Li X, Wang YF, Hao DM, Qiu CS, Chen XB (2011) Isolation and characterization of polymorphic microsatellite markers from flax (Linum usitatissimum L.). Afr J Biotechnol 10:734–739
Doligez A, Adam-Blondon AF, Cipriani G, Di Gaspero G, Laucou V, Merdinoglu D, Meredith CP, Riaz S, Roux C, This P (2006) An integrated SSR map of grapevine based on five mapping populations. Theor Appl Genet 113:369–382
Everaert I, de Riek J, de Loose M, van Waes J, van Bockstaele E (2001) Most similar variety grouping for distinctness evaluation of flax and linseed (Linum usitatissimum L.) varieties by means of AFLP and morphological data. Plant Var Seeds 14:69–87
FAOSTAT (2014) Food and Agriculture Organization. http://faostat3.fao.org/home/index.html
Fu YB (2006) Redundancy and distinctness in flax germplasm as revealed by RAPD dissimilarity. Plant Genet Resour 4:117–124
Fu YB, Diederichsen A, Richards KW, Peterson G (2002a) Genetic diversity within a range of cultivars and landraces of flax (Linum usitatissimum L.) as revealed by RAPDs. Genetic Res Crop Evol 49:167–174
Fu YB, Peterson G, Diederichsen A, Richards KW (2002b) RAPD analysis of genetic relationships of seven flax species in the genus Linum L. Genet Res Crop Evol 49:253–259
Gill KS (1987) Linseed publications and information division. Indian Council of Agricultural Research, New Delhi
Hackett CA, Broadfoot LB (2003) Effect of genotyping errors, missing values and segregation distortion in molecular marker data on the construction of linkage maps. Heredity 90:33–38
Hong Y, Chen X, Liang X, Liu H, Zhou G, Li S, Wen S, Holbrook CC, Guo B (2010) A SSR-based composite genetic linkage map for the cultivated peanut (Arachis hypogaea L.) genome. BMC Plant Biol 10:17
Kosambi DD (1994) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kumar S, You FM, Duguid S, Booker H, Rowland G, Cloutier S (2015) QTL for fatty acid composition and yield in linseed (Linum usitatissimum L.). Theor Appl Genet 128:965–984
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lu H, Romero-Severson J, Bernardo R (2002) Chromosomal regions associated with segregation distortion in maize. Theor Appl Genet 105:622–628
Oh TJ, Gorman M, Cullis CA (2000) RFLP and RAPD mapping in flax (Linum usitatissimum). Theor Appl Genet 101:590–593
Pali V, Verma SK, Xalxo MS, Saxena RR, Mehta N, Verulkar SB (2014) Identification of microsatellite markers for fingerprinting popular Indian flax (Linum usitatissimum L.) cultivars and their utilization in seed genetic purity assessments. Aust J Crop Sci 8:119
Pali V, Mehta N, Verulkar SB, Xalxo MS, Saxena RR (2015) Molecular diversity in Flax (Linum usitatissimum L.) as revealed by DNA based markers. Int J Plant Res 28:157–165
Roose-Amsaleg C, Cariou-Pham E, Vautrin D, Tavernier R, Solignac M (2006) Polymorphic microsatellite loci in Linum usitatissimum. Mol Ecol Notes 6:796–799
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNAsepacer-length polymorphism in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci (USA) 81:1757–1761
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Song KM, Suzuki JY, Slocum MK, Williams PM, Osborn TC (1991) A linkage map of Brassica rapa (syn. campestris) based on restriction fragment length polymorphism loci. Theor Appl Genet 82:296–304
Soto-Cerda BJ, Carrasco RA, Aravena GA, Urbina HA, Navarro CS (2011a) Identifying novel polymorphic microsatellites from cultivated flax (Linum usitatissimum L.) following data mining. Plant Mol Biol Rep 29:753–759
Soto-Cerda BJ, Saavedra HU, Navarro CN, Ortega PM (2011b) Characterization of novel genic SSR markers in Linum usitatissimum (L.) and their transferability across eleven Linum species. Electron J Biotechnol 14:4
Soto-Cerda BJ, Duguid S, Booker H, Rowland G, Diederichsen A, Cloutier S (2014) Association mapping of seed quality traits using the Canadian flax (Linum usitatissimum L.) core collection. Theor Appl Genet 127:881–896
Spielmeyer W, Green AG, Bittisnich D, Mendham N, Lagudah ES (1998) Identification of quantitative trait loci contributing to Fusarium wilt resistance on an AFLP linkage map of flax. Theor Appl Genet 97:633–641
Thoquet P, Gherardi M, Journet EP, Kereszt A, Ane JM, Prosperi JM, Huguet T (2002) The molecular genetic linkage map of the model legume Medicago truncatula: an essential tool for comparative legume genomics and the isolation of agronomically important genes. BMC Plant Biol 2:1
Uysal H, Fu Y-B, Kurt O, Peterson GW, Diederichsen A, Kusters P (2010) Genetic diversity of cultivated flax (Linum usitatissimum L.) and its wild progenitor pale flax (Linum bienne Mill.) as revealed by ISSR markers. Genet Resour Crop Evol 57:1109–1119
Voorrips RE (2006) MapChart 2.2: software for the graphical presentation of linkage maps and QTLs. Plant Research International, Wageningen
Wang S, Batsen CJ, Zeng ZB (2007) Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh
Westcott ND, Muir ND (2003) Chemical studies on the constituents of Linum sp. In: Muir AD, Westcott ND (eds) Flax, the genus Linum. Taylor and Francis, New York, pp 55–73
Acknowledgements
Authors thank the Director, CSIR-NBRI, Lucknow for providing the facilities to carry out the present investigation. Financial support in form of DST-INSPIRE Fellowship to Chandrawati is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chandrawati, Yadav, H.K. Development of linkage map and mapping of QTLs for oil content and yield attributes in linseed (Linum usitatissimum L.). Euphytica 213, 258 (2017). https://doi.org/10.1007/s10681-017-2050-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-2050-x