
Abstract
Flowering represents the transition from the vegetative to reproductive phase and plays an important role in many agronomic traits. For soybean, a short day (SD) induced and photoperiod-sensitive plant, delaying flowering time under SD environments is very important and has been used by breeders to increase yields and enhance plant adaptabilities at lower latitudes. The purpose of this study was to identify quantitative trait loci (QTLs) associated with flowering time, especially QTLs underlying the long juvenile (LJ) trait which delays flowering time under SD environments. A population of 91 recombinant inbred lines derived from a cross between AGS292 and K3 was used for map construction and QTL analysis. The map covered 2546.7 cM and included 52 new promoter-specific indel and 9 new exon-specific indel markers. The phenotypic days-to-flowering data were examined in nine environments, including four short-day (SD, low latitude) and five long-day photoperiod (LD, high latitude) environments. For the SD environments, six QTLs were detected. Five of them were associated with the LJ trait. Among the five LJ QTLs, four QTLs may be attributed to the known flowering time genes, including qFT-J-1 for FT5a locus, qFT-J-2 for the FT2a locus, qFT-O for the E2 locus and qFT-L for the E3 locus. This is the first report that the E2, E3, FT2a and FT5a loci may be associated with the LJ trait. Under the five LD environments, as expected, qFT-O for the E2 locus and qFT-L for the E3 locus were identified, suggesting that E2 and E3 loci are very important for soybean adaptation in LD photoperiod. Conjoint analysis of multiple environments identified nine additive QTLs and nine pairs of epistatic QTLs, among which most were involved in interactions with the environments. In total, five QTLs (qFT-B2-1, qFT-C1-1, qFT-K, qFT-D2 and qFT-F) were identified that may represent novel flowering time genes. This provides a fundamental foundation for future studies of flowering time in soybean using fine mapping, map-based cloning, and molecular-assisted breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flowering represents the transition from the vegetative to reproductive phase in plants and is influenced by many factors (Levy and Dean 1998). One of the important cue is the photoperiod. Soybean [Glycine max (L.) Merr.] is sensitive to photoperiod, which makes each cultivar is restricted to a very narrow range of latitudes (Pooprompan et al. 2006). Widely adaptable soybean cultivars have been created by natural variation in the major genes and quantitative trait loci (QTLs) controlling flowering. By classic methods, ten major genes (E1-E9, and J) controlling flowering and maturity time have been characterized in soybean (Bernard 1971; Buzzell 1971; Buzzell and Voldeng 1980; McBlain and Bernard 1987; Ray et al. 1995; Bonato and Vello 1999; Cober and Voldeng 2001a, b; Cober and Morrison 2010; Kong et al. 2014). Among these genes, E1 has been cloned by a map-based approach and identified as a legume-specific transcription factor with a putative nuclear localization signal and a domain distantly related to the B3 domain (Xia et al. 2012), and E2 has been identified as a soybean ortholog of the Arabidopsis GIGANTEA gene (Watanabe et al. 2011). E3 has been confirmed as a phyA homolog by fine-mapping around a QTL for flowering time (qFT3) (Watanabe et al. 2009). Liu et al. (2008) have concluded that the E4 gene also encodes a soybean phyA protein and that the recessive e4 allele is a loss-of-function allele caused by the insertion of a Ty1/copia-like retrotransposon. In cultivated soybean, there are at least three mutated alleles in the E1 gene (Xia et al. 2012), four in the E3 gene (Xu et al. 2013) and six in the E4 gene (Tsubokura et al. 2013). The diversity of the allelic variations and the different allelic combinations of the E1, E3 and E4 genes condition soybean flowering time, post-flowering responses and photoperiod insensitivity and greatly contribute to the wide adaptation of soybean (Xu et al. 2013; Jiang et al. 2014). In addition to these cloned maturity genes, among the more than ten copies of the FLOWERING LOCUS T (FT) homolog in the soybean genome, two homologs, GmFT2a and GmFT5a, have been found to encode components of ‘‘florigen’’, the mobile flowering promotion signal that is involved in the transition to flowering, and these two FT homologs coordinately control flowering in soybean (Kong et al. 2010). GmFT2a and GmFT5a redundantly and differentially regulate flowering through interactions with the bZIP transcription factor, GmFDL19, for the subsequent up-regulation of this protein in soybean (Nan et al. 2014). The E1, E2, E3 and E4 maturity genes have been shown to down-regulate GmFT2a and GmFT5a expression to delay flowering and maturation under LD conditions in soybean, suggesting that GmFT2a and GmFT5a are the soybean flowering integrators and major flowering regulation targets (Kong et al. 2010; Thakare et al. 2011; Watanabe et al. 2011).
In previous research, the genes mentioned above (E1, E2, E3, E4, GmFT2a and GmFT5a) were shown to play an important role only in LD photoperiod. It is known that soybean is a short-day (SD) plant, and most cultivars have a SD requirement for floral induction. When soybean cultivars are grown under SD conditions, cultivars with sensitivity to photoperiod flower early, result in low grain yield, and consequently limit the growing area. It is therefore important to research the genetic control on delaying flowering time under SD environments. This trait was termed the “long-juvenile” (LJ) trait (Parvez and Gardner 1987; Sinclair and Hinson 1992; Ray et al. 1995). The LJ trait plays a pivotal role in extending the range of adaptation of soybean cultivars to lower latitudes and to new management schemes with shifted sowing dates in tropical countries. It has been reported that the northward expansion of soybean production in South America, where more extensive research has been performed, is dependent on the LJ trait (Spehar 1995). However, the genetic control mechanism for this trait remains elusive. Two genes, J and E6, had been reported to play important role in LJ trait (Ray et al. 1995; Bonato and Vello 1999). The single locus J has been identified in a number of crosses with PI 159925 (Ray et al. 1995). The single locus E6 is created by natural variation in ‘Paraná’, and finally produces the long-juvenile ‘Paranagoiana’ (Bonato and Vello 1999). Recently, an F2 population resulting from a cross between conventional juvenile (CJ) lines OT94-47 and the LJ line Paranagoiana exhibited a 15:1 early:late flowering ratio in 12 h photoperiods. A similar 15:1 ratio was observed in offspring of a cross between CJ line OT94-47 and the LJ line PI 159925. These results suggest that the LJ trait is conditioned by at least two recessive alleles in PI 159925 and Paranagoiana (Cober 2011). Further studies of LJ parents have shown that recessive alleles at two or three loci control the long-juvenile trait (Carpentieri-Pípolo et al. 2000, 2002). Though so many researched had been conducted on LJ trait, but only one gene, J, has been mapped to the soybean linkage group Gm 04 between the SSR markers Sat_337 and Satt396, where the genetic distance between the J allele and the closet marker Sat_337 is 0.7 cM (Cairo et al. 2002, 2009).
In addition to these major genes, many QTLs controlling flowering time have been reported (Keim et al. 1990; Lee et al. 1996; Tasma et al. 2001; Chapman et al. 2003; Funatsuki et al. 2005; Liu et al. 2007; Khan et al. 2008; Liu and Abe 2010; Cheng et al. 2011). Some of these QTLs most likely correspond with one of the known major genes, such as E1, E2, E3, E4, or E8 (Watanabe et al. 2004; Funatsuki et al. 2005; Githiri et al. 2007; Khan et al. 2008; Liu and Abe 2010; Cheng et al. 2011), while the others are described in the SoyBase database (http://soybase.org/). In addition to affecting flowering and maturity, the major genes and QTLs for flowering often influence agronomic traits, including plant height and yield (Lee et al. 1996; Chapman et al. 2003; Cober and Morrison 2010), degree of cleistogamy (Khan et al. 2008), seed coat pigmentation, and cracking caused by chilling stress (Takahashi and Abe 1999; Githiri et al. 2007). Therefore, the understanding of QTLs at the molecular level and their interactions with environmental factors will help to optimize the genotypic combinations that lead to higher or more stable yields during the cropping season in a particular region.
The objectives of the present study were as follows: (1) to identify QTLs associated with soybean flowering time using a recombinant inbred line (RIL) population exposed to different environments; (2) to identify QTLs associated with the LJ trait under different SD environments; and (3) to analyze the interactions between QTLs and the environments.
Materials and methods
Plant materials
A population of 91 F9 soybean RILs obtained by single seed descent (SSD) from a cross between AGS292 and K3 was used. The vegetable soybean cultivar AGS292 was a pure line selected from the Japanese cultivar ‘Taishoshiroge’ by the AVRDC (the World Vegetable Center, Taiwan). K3 was a grain soybean that delayed flowering than AGS292. It was a pure line derived by pedigree selection from a cross between ‘G8891’ and ‘G7945’ (both were obtained from the AVRDC collection) by the soybean breeding project of Kasetsart University, Thailand.
Field observation
Seeds from each RIL and the parents were planted at Kasetsart University, Kamphaeng Saen Campus, Nakhon Pathom Province, Thailand (13°82′N, 100°04′E). Field trials were carried out over two seasons (rainy and dry) and two years (August 2004–February 2005 and August 2010–February 2011). The plot was located between Equator and the Tropic of Cancer, where belonged to low latitudes, so is considered a SD environment.
The RILs were also grown under LD conditions in Japan and China. In Japan, seeds were sown in June of 2010 and 2011 in the research field of the National Institute of Agrobiological Sciences at Tsukuba (36°02′N, 140°11′E) and in May of 2010 in the field of Hokkaido University, Sapporo (43°07′N, 141°39′E). In China, the seeds were sown in May of 2010 in the field of the Northeast Institute of Geography and Agroecology, Chinese Academy of Sciences, Harbin (45°44′N, 126°36′E) and in June of 2011 in the field of Shandong Normal University at Jinan (36°40′N, 117°00′E). These plots were located north of the Tropic of Cancer, where belonged to mid-latitude regions, so were considered LD environments.
In total, the QTLs were analyzed in nine different environments. On each of the nine experimental occasions, all 91 lines, together with their parents AGS292 and K3, were grown in three fully randomized block replications. Every block contained all 91 lines and parents. Each individual was sampled for analysis of the phenotypic parameter flowering time (R1), which was defined as the time from emergence to the opening of the first flower (Fehr et al. 1971). Flowering times were tested for deviations from normality using the parameters of kurtosis and skewness by SPSS 16.0 software (SPSS Inc., Chicago, IL, USA).
DNA isolation and molecular marker analysis
DNA was extracted from the young leaves of each RIL and the parents following a previously described method (Doyle et al. 1990). SSR analysis was built on using primers selected from an integrated soybean genetic linkage map (Cregan et al. 1999; Song et al. 2004; Hyten et al. 2010). The SSR primer sequences were obtained from the SoyBase web site of the USDA, ARS Soybean Genome Database (http://soybase.agron.iastate.edu/). In addition, we developed 52 promoter-specific indel (PSI) and 9 exon-specific indel (ESI) markers (Table S1). Five allele-specific markers for E2 (Watanabe et al. 2011), E3 (Xu et al. 2013), E4 (Liu et al. 2008), FT2a and FT3a (Kong et al. 2010) were also used. The polymerase chain reaction (PCR) mixture contained 30 ng of total genomic DNA, 0.25 µM of 5′ and 3′ primers, 200 µM of each dNTP, 0.5 U of Taq polymerase (TaKaRa, Otsu, Japan) and 1 × PCR buffer (10 mM Tris–HCl, pH 8.3, 50 mM KCl, and 1.5 mM MgCl2) in a total volume of 20 µL. PCR was performed with a GeneAmp PCR System 9700 (Perkin Elmer/Applied Biosystems, Foster City, CA, USA) using the following program: 94 °C for 5 min, followed by 35 cycles of 30 s at 94 °C, 30 s at 48 °C, and 30 s at 72 °C, and a final step of 5 min at 72 °C. PCR products were separated on a 6 % denatured polyacrylamide gel (PAGE) by electrophoresis.
Genetic linkage map construction
In total, 338 polymorphic and informative markers, including 52 PSI, 9 ESI, 5 allele-specific and 272 SSR markers, were chosen as anchors to construct the linkage map covering all 20 linkage groups. Marker order and distance were determined by Map Manager program QTXb20 ( http://mapmgr.roswellpark.org/mapmgr.html) using the Kosambi function and a criterion of 0.001 probability (d.f. = 1). Most of the markers were assigned to the 20 linkage groups as expected from the integrated map (Cregan et al. 1999; Song et al. 2004). Finally, we used Mapchart 2.1 to draw the linkage groups (Voorrips 2002).
Statistical analysis and QTL identification
Two models were used to detect QTLs and analyze the interactions between the QTLs and the environments: the multiple QTL model (MQM), implemented by MapQTL 5.0 (Van Ooijen 2004), and mixed linear-based composite interval mapping (MCIM), implemented by QTLNetwork 2.1 (Yang et al. 2008).
For the MQM, a LOD score of 3.0 was used as a minimum to declare the significance of a QTL in a particular genomic region. 1000 permutations at a 0.05 probability were also conducted to identify the genome-wide LOD (Churchill and Doerge 1994). QTLs with a LOD score exceeding the genome-wide LOD were declared as significant QTLs, whereas the other QTLs with LOD less than the genome-wide LOD but more than 3.0 were identified as suggestive QTLs.
MCIM was used to map QTLs with additive and epistatic effects as well as their interactions with the environments (additive by environment and epistatic by environment). This analysis was performed using a 2D genome scan, with a 1-cM walking speed and 10-cM window size. Significant thresholds (critical F-values) for QTL detection were calculated with 1000 permutations and a genome-wide error rate of 0.05.
Results
Phenotypic analysis
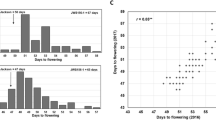
For different environments, the average flowering time for each RIL was used to analyze the segregation pattern (Table 1). We found that the skewness and kurtosis values of different environments deviated slightly from zero, except for the 2010 dry season in Thailand. These results show that the segregation pattern of this trait under different environments fits the normal distribution model and the RILs can be used for genetic map construction and QTL identification. The RILs under LD conditions flowered significantly later than those under SD conditions. The flowering time of the RILs grown at Harbin was the longest of the nine environments, which may be attributed to its high latitude (45°N).
Construction of genetic linkage map
Using polymorphic 338 markers, a genetic linkage map covering 2546.7 cM was constructed using the Kosambi function (Figure S1). The main marker type contributing to this linkage map was the SSR markers, while the linkage gaps between the SSR markers were bridged by indel PSI and ESI markers. However, the Gm 18 chromosome still lacked polymorphic markers and was divided into Gm 18-1 and Gm 18-2. The map length is approximately consistent with the currently known recombination distance of 2524 cM in the integrated soybean linkage map (Cregan et al. 1999; Song et al. 2004). The marker order of our map was in good accordance with that of the integrated map with only slight differences. However, all of the discordant marker orders occurred within 5 cM of their respective orders on the integrated map.
QTL identification of LJ trait under SD conditions
Under the four SD environments, a total of six QTLs was detected by the MQM (Table 2). They were distributed over four linkage groups and explained 15.2–35.4 % of the phenotypic variation. The additive effect for qFT-F was positive, which indicated that the positive allele for this QTL originated from AGS292. The other five QTLs originated from K3, i.e. they delayed flowering time under SD and were associated with the LJ trait. Among the six QTLs, only qFT-J-2 significantly (p < 0.05) affected flowering time as shown by genome-wide analyses with permutation tests for two rainy environments. It accounted for 34.4 and 35.4 % of the total variances observed for the two environments (Table 2). When we used MCIM to detect QTLs for single SD environment at a 0.001 significant probability level, only qFT-J-2 and qFT-O were detected, and the other four were missed (Table S2). qFT-J-2 was consistently detected under different SD environments by both MCIM and MQM approaches, suggesting that it is the major QTL conditioning LJ trait in this RIL population.
QTL identification of flowering time under LD conditions
Under the five LD environments, two QTLs, qFT-O and qFT-L, were identified by the MQM (Table 2). They were located near the allele-specific markers for E2 and E3, respectively, and explained 18.1–50.5 % of the phenotypic variance, with additive effects ranging from 4.32 to 12.15, suggesting that these two QTLs may be attributed to the E2 and E3 loci. Either the qFT-L or the E3 locus affected flowering time as shown by genome-wide analyses of all five LD environments with permutation tests. When we detected QTLs by MCIM at a 0.001 significant probability level, in addition to qFT-O and q-FT-L, qFT-I was also identified. qFT-I was located near the allele-specific markers for E4 suggesting that qFT-I may be conferred by the E4 locus; it was found to exist in four LD environments except at Jinan in 2011 (Table S2). Using allele-specific markers of E1, E2, E3 and E4 genes, the genotypes at these four loci of the two parents AGS292 and K3 were identified as E1e2e3e4 and E1E2E3E4, respectively. The genotyping results confirmed that flowering QTLs qFT-O, qFT-L and qFT-I were conditioned by E2, E3 and E4 genes, respectively. Our results also suggest that the two approaches for detecting QTL, MCIM and MQM, can complement each other to pyramid QTLs in RIL population. qFT-I had an epistatic effect with qFT-L in four LD environments (Table S3). This epistasis contributed 2.45–8.58 days to the flowering time and accounted for 5.63–12.09 % of the phenotypic variance.
QTLs with additive and additive-by-environment interaction effects under nine environments
In order to analyze the interactions between QTLs and environments, we performed a conjoint analysis. Compared with the single environment analysis, we detected four additional minor QTLs: qFT-B2-1, qFT-C1-1, qFT-D2 and qFT-J-4 (Table 3). These four QTLs demonstrated weak interactions with the environment. The other five QTLs, which were also detected in the single environment analysis, displayed additive-by-environment interaction effects with multiple environments. These additive-by-environment interaction effects were opposite between the LD and SD environments, which indicated that the environments had different roles on the genes for these QTLs (LD and SD). Of the nine QTLs, the qFT-L or E3 locus was responsible for the largest phenotypic variation due to both additive and additive-by-environment effects, and the heritability of the additive effect was higher than that of the additive-by-environment effect, which showed that genotypic background had a greater effect on this QTL than the environment.
QTLs with epistasis and epistasis-by-environment interaction effects for nine environments
Nine pairs of QTLs with epistatic effects were detected (Table 4). Among these effects, the epistasis occurring between qFT-I and qFT-L was the largest, contributing 2.26 days to the delayed flowering time and accounting for 1.96 % of the phenotypic variance by epistasis in multiple environments. This epistasis also had significant interaction effects with five environments (p < 0.001) (Table 4). We detected three other QTLs by epistatic effects only: qFT-B2-2, qFT-K and qFT-C1-2. These results indicate that analysis of the interactions between the environment and the QTLs allowed for the detection of more minor QTLs.
Discussion
QTLs for LJ trait
Delayed flowering and maturity time under SD conditions in soybean was termed the LJ trait (Hartwig and Kiihl 1979; Ray et al. 1995; Spehar 1995). This trait is especially important for extending the range of adaptation of soybean to lower latitudes and to new management schemes with shifted sowing dates to increase soybean productivity in such regions (Hartwig and Kiihl 1979; Ray et al. 1995; Spehar 1995). To date, there are few reports of the detection of LJ QTLs through multiple environments using RILs (Liu et al. 2011). In our study, we grew RILs under four SD environments and identified five QTLs for the LJ trait, including qFT-O, qFT-J-1, qFT-J-2, qFT-J-3 and qFT-L, in which all the alleles originating from K3 delayed flowering time and were considered to condition the LJ trait. Among the five LJ QTLs, qFT-O, qFT-J-1, qFT-J-2 and qFT-L were localized to the regions near the allele–specific DNA markers for E2, GmFT5a, GmFT2a and E3, respectively (Watanabe et al. 2009; Kong et al. 2010; Watanabe et al. 2011). Previous research suggests that maturity genes E2, E3 and E4 do not have any effect on flowering time and maturity under SD conditions (Cober et al. 1996). Surprisingly, we found that qFT-O (E2 gene) and qFT-L (E3 gene) can be detected under a SD environment and in association with the LJ trait (Table 2). To our knowledge, this is the first report that the E2 and E3 genes condition flowering time (or the LJ trait) under SD conditions. In addition, while in other genetic models the recessive allele conditioned the LJ trait (Carpentieri-Pípolo et al. 2000, 2002), the dominant allele from the E2 and E3 loci conditioned the LJ trait in our study. Further study is needed to confirm this new finding. qFT-J-1 and qFT-J-2 mapped very tightly to allele-specific markers of GmFT5a and GmFT2a, the two florigens of soybean (Kong et al. 2010), suggesting that GmFT5a and GmFT2a may condition the LJ trait in soybean.
To minimum the influence of environmental factors affecting flowering time of the LJ trait, the 91 RILs and the parental lines were grown at 25 °C under SD conditions (12L/12D) with three replications in growth chambers. Any three of the seeds were grown in one plant pot and all of the plant pots were randomly placed. The flowering time for every seed was detected. The phenotypic data for them can find in table S4. The four QTLs qFT-J-1, qFT-J-2, qFT-J-3 and qFT-L could also be detected by the MQM (Table S5). These results confirm that these four QTLs, particularly those located in association with the E3 locus, were truly present under a SD environment in both indoor and outdoor conditions. The interval for qFT-J-2 and qFT-J-3 had already been found to be associated with flowering time in previous studies (Tasma et al. 2001; Pooprompan et al. 2006). It will be of great interest to perform fine mapping to further elucidate the underlying genetic mechanisms of these QTLs.
Relationships between QTLs and the environments
The results of single environment analysis do not always provide valid predictions of the effects of QTLs controlling a target trait. Analysis by MCIM has been proven to be effective for detecting minor-effect QTLs in a variety of crops (Wang et al. 1999; Gutierrez-Gonzalez et al. 2009, 2010; Xu et al. 2014). In the present study, we used multiple environments to perform an integrated analysis by MCIM, identifying nine additive QTLs. Compared with single environment analysis, four additional QTLs (qFT-B2-1, qFT-C1-1, qFT-D2 and qFT-J-4) were detected and had little interactions with the environments. qFT-B2-1 was located near the marker Satt467. In the SoyBase database (http://soybase.org/), there was only one QTL for flowering time near the marker Satt534 on Gm 14. Compared with the integrated soybean linkage map, Satt467 is located at 19.17 cM and Satt534 is located at 75.73 cM (Hyten et al. 2010). They were far from each other, therefore, qFT-B2-1 may be a new QTL for flowering time (Reinprecht et al. 2006).
Using multiple environments to perform integrated analysis by MCIM not only greatly facilitated the detection of QTLs but also allowed for the identification of epistatic and epistasis-by-environment interaction effects. These results further elucidate the mechanisms underlying the genetic control of flowering time. In this study, three QTLs (qFT-B2-2, qFT-K and qFT-C1-2) were detected with only epistatic effects (Table 4). qFT-C1-2 was located between Sat_140 and GMES0780. This interval was very close to the marker Sat_337, which harbored the J allele that is associated with the LJ trait (Cairo et al. 2009). Thus, it was clear that analysis of interactions between the QTLs and the environments facilitated the detection of QTLs. None of these QTLs had major effects, but they were able to influence flowering time through interactions with other loci, an observation in accordance with those reported by Jannink (2007). Furthermore, for qFT-K, no QTL associated with flowering has been previously identified (Li et al. 2010; Ha et al. 2012).
Conclusions
The objective of this study was to identify QTLs associated with flowering time, especially for the LJ trait. Under SD environments, we identified a total of six QTLs. Of these, qFT-F has not been previously reported, suggesting that it is a novel QTL for flowering time. The other five QTLs originated form K3 and were associated with the LJ trait. Among the five LJ QTLs, four QTLs, qFT-J-1, qFT-J-2, qFT-O and qFT-L, may control the known genes GmFT5a, GmFT2a, E2 and E3, and this is the first report that these genes may be associated with the LJ trait. Additional studies are necessary to confirm these new findings. In addition, we also identified five QTLs (qFT-B2-1, qFT-C1-1, qFT-K, qFT-D2 and qFT-F) by the integrated analysis, which may represent novel flowering time genes.
In conclusion, our research provides insights into the mechanisms of flowering time, especially with regard to the LJ trait. The information obtained from our findings will facilitate gene cloning and functional elucidation for soybean molecular breeding under different environmental conditions.
References
Bernard R (1971) Two major genes for time of flowering and maturity in soybeans. Crop Sci 11:242–244
Bonato ER, Vello NA (1999) E6, a dominant gene conditioning early flowering and maturity in soybeans. Genet Mol Biol 22:229–232
Buzzell R (1971) Inheritance of a soybean flowering response to fluorescent-daylength conditions. Can J Genet Cytol 13:703–707
Buzzell R, Voldeng H (1980) Inheritance of insensitivity to long daylength. Soybean Genet Newsl 7:26–29
Cairo CA, Stein J, Delgado L, Bortolotti S, Guelman SA, Ortiz JPA, Morandi EN (2002) Tagging the juvenile locus in soybean [Glycine max (L.) Merr.] with molecular markers. Euphytica 124:387–395
Cairo CA, Cambursano MV, Morand EN (2009) Molecular mapping of the juvenile locus in soybean. In: World soybean research conference VIII, Beijing, China, 10–15 August 2009
Carpentieri-Pípolo V, de Almeida LA, de Souza Kiihl RA, Rosolem CA (2000) Inheritance of long juvenile period under short day conditions for the BR80-6778 soybean (Glycine max (L.) Merrill) line. Euphytica 112:203–209
Carpentieri-Pípolo V, Almeida LAd, Kiihl RAdS (2002) Inheritance of a long juvenile period under short-day conditions in soybean. Genet Mol Biol 25:463–469
Chapman A, Pantalone V, Ustun A, Allen F, Landau-Ellis D, Trigiano R, Gresshoff P (2003) Quantitative trait loci for agronomic and seed quality traits in an F2 and F4: 6 soybean population. Euphytica 129:387–393
Cheng L, Wang Y, Zhang C, Wu C, Xu J, Zhu H, Leng J, Bai Y, Guan R, Hou W (2011) Genetic analysis and QTL detection of reproductive period and post-flowering photoperiod responses in soybean. Theor Appl Genet 123:421–429
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Cober ER (2011) Long juvenile soybean flowering responses under very short photoperiods. Crop Sci 51:140–145
Cober ER, Morrison MJ (2010) Regulation of seed yield and agronomic characters by photoperiod sensitivity and growth habit genes in soybean. Theor Appl Genet 120:1005–1012
Cober ER, Voldeng HD (2001a) Low R:FR light quality delays flowering of soybean lines. Crop Sci 41:1823–1826
Cober ER, Voldeng HD (2001b) A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T. Crop Sci 41:698–701
Cober E, Tanner J, Voldeng H (1996) Soybean photoperiod-sensitivity loci respond differentially to light quality. Crop Sci 36:606–610
Cregan P, Jarvik T, Bush A, Shoemaker R, Lark K, Kahler A, Kaya N, VanToai T, Lohnes D, Chung J (1999) An integrated genetic linkage map of the soybean genome. Crop Sci 39:1464–1490
Doyle JJ, Doyle JL, Brown A (1990) A chloroplast-DNA phylogeny of the wild perennial relatives of soybean (Glycine subgenus Glycine): congruence with morphological and crossing groups. Evolution 44:371–389
Fehr W, Caviness C, Burmood D, Pennington J (1971) Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci 11:929–931
Funatsuki H, Kawaguchi K, Matsuba S, Sato Y, Ishimoto M (2005) Mapping of QTL associated with chilling tolerance during reproductive growth in soybean. Theor Appl Genet 111:851–861
Githiri SM, Yang D, Khan NA, Xu D, Komatsuda T, Takahashi R (2007) QTL analysis of low temperature–induced browning in soybean seed coats. J Hered 98:360–366
Gutierrez-Gonzalez JJ, Wu X, Zhang J, Lee J-D, Ellersieck M, Shannon JG, Yu O, Nguyen HT, Sleper DA (2009) Genetic control of soybean seed isoflavone content: importance of statistical model and epistasis in complex traits. Theor Appl Genet 119:1069–1083
Gutierrez-Gonzalez JJ, Wu X, Gillman JD, Lee J-D, Zhong R, Yu O, Shannon G, Ellersieck M, Nguyen HT, Sleper DA (2010) Research article Intricate environment-modulated genetic networks control isoflavone accumulation in soybean seeds. BMC Plant Biol 10:105
Ha B-K, Kim H-K, Kang S-T (2012) Mapping QTLs with epistatic effects and QTL-by-environment interactions for seed coat cracking in soybeans. Euphytica 186:933–942
Hartwig EE, Kiihl RA (1979) Identification and utilization of a delayed flowering character in soybeans for short-day conditions. Field Crops Res 2:145–151
Hyten DL, Choi I-Y, Song Q, Specht JE, Carter TE, Shoemaker RC, Hwang E-Y, Matukumalli LK, Cregan PB (2010) A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping. Crop Sci 50:960–968
Jannink J-L (2007) Identifying quantitative trait locus by genetic background interactions in association studies. Genetics 176:553–561
Jiang B, Nan H, Gao Y, Tang L, Yue Y, Lu S, Ma L, Cao D, Sun S, Wang J, Wu C, Yuan X, Hou W, Kong F, Han T, Liu B (2014) Allelic combinations of soybean maturity loci E1, E2, E3 and E4 result in diversity of maturity and adaptation to different latitudes. PLoS One 9:e106042
Keim P, Diers B, Olson T, Shoemaker R (1990) RFLP mapping in soybean: association between marker loci and variation in quantitative traits. Genetics 126:735–742
Khan NA, Githiri SM, Benitez ER, Abe J, Kawasaki S, Hayashi T, Takahashi R (2008) QTL analysis of cleistogamy in soybean. Theor Appl Genet 117:479–487
Kong F, Liu B, Xia Z, Sato S, Kim BM, Watanabe S, Yamada T, Tabata S, Kanazawa A, Harada K (2010) Two coordinately regulated homologs of FLOWERING LOCUS T are involved in the control of photoperiodic flowering in soybean. Plant Physiol 154:1220–1231
Kong F, Nan H, Cao D, Li Y, Wu F, Wang J, Lu S, Yuan X, Cober ER, Abe J (2014) A new dominant gene conditions early flowering and maturity in soybean. Crop Sci 54:2529–2535
Lee S, Bailey M, Mian M, Shipe E, Ashley D, Parrott W, Hussey R, Boerma H (1996) Identification of quantitative trait loci for plant height, lodging, and maturity in a soybean population segregating for growth habit. Theor Appl Genet 92:516–523
Levy YY, Dean C (1998) The transition to flowering. Plant Cell 10:1973–1989
Li H, Liu H, Han Y, Wu X, Teng W, Liu G, Li W (2010) Identification of QTL underlying vitamin E contents in soybean seed among multiple environments. Theor Appl Genet 120:1405–1413
Liu B, Abe J (2010) QTL mapping for photoperiod insensitivity of a Japanese soybean landrace Sakamotowase. J Hered 101:251–256
Liu B, Fujita T, Yan Z-H, Sakamoto S, Xu D, Abe J (2007) QTL mapping of domestication-related traits in soybean (Glycine max). Ann Bot 100:1027–1038
Liu B, Kanazawa A, Matsumura H, Takahashi R, Harada K, Abe J (2008) Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A gene. Genetics 180:995–1007
Liu W, Kim MY, Kang YJ, Van K, Lee YH, Srinives P, Yuan DL, Lee SH (2011) QTL identification of flowering time at three different latitudes reveals homelogous genomic regions that control flowering in soybean. Theor Appl Genet 123:545–553
McBlain B, Bernard R (1987) A new gene affecting the time of flowering and maturity in soybeans. J Hered 78:160–162
Nan H, Cao D, Zhang D, Li Y, Lu S, Tang L, Yuan X, Liu B, Kong F (2014) GmFT2a and GmFT5a redundantly and differentially regulate flowering through interaction with and upregulation of the bzip transcription factor GmFDL19 in soybean. PLoS One 9:e97669
Parvez A, Gardner F (1987) Daylength and sowing date responses of soybean lines with “juvenile” trait. Crop Sci 27:305–310
Pooprompan P, Wasee S, Toojinda T, Abe J, Chanprame S, Srinives P (2006) Molecular marker analysis of days to flowering in vegetable soybean (Glycine max (L.) Merrill). Kasetsart J Nat Sci 40:573–581
Ray JD, Hinson K, Mankono J, Malo MF (1995) Genetic control of a long-juvenile trait in soybean. Crop Sci 35:1001–1006
Reinprecht Y, Poysa VW, Yu K, Rajcan I, Ablett GR, Pauls KP (2006) Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 49:1510–1527
Sinclair TR, Hinson K (1992) Soybean flowering in response to the long-juvenile trait. Crop Sci 32:1242–1248
Song Q, Marek L, Shoemaker R, Lark K, Concibido V, Delannay X, Specht JE, Cregan P (2004) A new integrated genetic linkage map of the soybean. Theor Appl Genet 109:122–128
Spehar CR (1995) Impact of strategic genes in soybean on agricultural development in the Brazilian tropical savannahs. Field Crops Res 41:141–146
Takahashi R, Abe J (1999) Soybean maturity genes associated with seed coat pigmentation and cracking in response to low temperatures. Crop Sci 39:1657–1662
Tasma I, Lorenzen L, Green D, Shoemaker R (2001) Mapping genetic loci for flowering time, maturity, and photoperiod insensitivity in soybean. Mol Breed 8:25–35
Thakare D, Kumudini S, Dinkins RD (2011) The alleles at the E1 locus impact the expression pattern of two soybean FT-like genes shown to induce flowering in Arabidopsis. Planta 234:933–943
Tsubokura Y, Matsumura H, Xu M, Liu B, Nakashima H, Anai T, Kong F, Yuan X, Kanamori H, Katayose Y (2013) Genetic variation in soybean at the maturity locus E4 is involved in adaptation to long days at high latitudes. Agronomy 3:117–134
Van Ooijen J (2004) MapQTL® 5. Software for the mapping of quantitative trait loci in experimental populations. Kyazma BV, Wageningen
Voorrips R (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang D, Zhu J, Li Z, Paterson A (1999) Mapping QTLs with epistatic effects and QTL × environment interactions by mixed linear model approaches. Theor Appl Genet 99:1255–1264
Watanabe S, Tajuddin T, Yamanaka N, Hayashi M, Harada K (2004) Analysis of QTLs for reproductive development and seed quality traits in soybean using recombinant inbred lines. Breed Sci 54:399–407
Watanabe S, Hideshima R, Xia Z, Tsubokura Y, Sato S, Nakamoto Y, Yamanaka N, Takahashi R, Ishimoto M, Anai T (2009) Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 182:1251–1262
Watanabe S, Xia Z, Hideshima R, Tsubokura Y, Sato S, Yamanaka N, Takahashi R, Anai T, Tabata S, Kitamura K (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 188:395–407
Xia Z, Watanabe S, Yamada T, Tsubokura Y, Nakashima H, Zhai H, Anai T, Sato S, Yamazaki T, Lü S (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci 109:E2155–E2164
Xu M, Xu Z, Liu B, Kong F, Tsubokura Y, Watanabe S, Xia Z, Harada K, Kanazawa A, Yamada T (2013) Genetic variation in four maturity genes affects photoperiod insensitivity and PHYA-regulated post-flowering responses of soybean. BMC Plant Biol 13:91
Xu Y, Wang R, Tong Y, Zhao H, Xie Q, Liu D, Zhang A, Li B, Xu H, An D (2014) Mapping QTLs for yield and nitrogen-related traits in wheat: influence of nitrogen and phosphorus fertilization on QTL expression. Theor Appl Genet 127:59–72
Yang J, Hu C, Hu H, Yu R, Xia Z, Ye X, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24:721–723
Acknowledgments
This work was funded by the National Natural Science Foundation of China (31430065, 31071445, 31171579, 31201222, 31230050, 31371651 and 31371643); the Open Foundation of the Key Laboratory of Soybean Molecular Design Breeding, Chinese Academy of Sciences; “Hundred Talents” Program of Chinese Academy of Sciences; Strategic Action Plan for Science and Technology Innovation of Chinese Academy of Sciences (XDA08030108); and Heilongjiang Natural Science Foundation of China (ZD201001, JC201313).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Sijia Lu, Ying Li, and Jialin Wang have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lu, S., Li, Y., Wang, J. et al. QTL mapping for flowering time in different latitude in soybean. Euphytica 206, 725–736 (2015). https://doi.org/10.1007/s10681-015-1501-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-015-1501-5