
Abstract
High-resolution diffuse reflectance spectra in the visible and near-infrared wavelengths were used to predict chemical properties of sediment samples obtained from Lake Okeechobee (FL, USA). Chemometric models yielded highly effective prediction (relative percent difference (RPD) = SD/RMSE >2) for some sediment properties including total magnesium (Mg), total calcium (Ca), total nitrogen (TN), total carbon (TC), and organic matter content (loss on ignition (LOI)). Predictions for iron (Fe), aluminum (Al), and various forms of phosphorus (total P (TP), HCl-extractable P (HCl-P), and KCl-extractable P (KCl-P)) were also sufficiently accurate (RPD > 1.5) to be considered useful; predictions for other P fractions as well as all pore water properties were poor. Notably, scanning wet sediments resulted in only a 7 % decline in RPD scores. Moreover, interpolation maps based on values predicted from wet sediment spectra captured the same spatial patterns for Ca, Mg, TC, TN, and TP as maps derived directly from wet chemistry, suggesting that field scanning of perpetually saturated sediments may be a viable option for expediting sample analysis and greatly reducing mapping costs. Indeed, the accuracy of spectral model predictions compared favorably with the accuracy of kriging model predictions derived from wet chemistry observations suggesting that, for some analytes, higher density spatial sampling enabled by use of field spectroscopy could increase the geographic accuracy of monitoring for changes in lake sediment chemical properties.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Environmental monitoring over large heterogeneous systems presents a significant logistical and budgetary challenge. In particular, traditional chemical analysis of soil/sediment samples is expensive and labor-intensive and presents a limiting factor affecting the resolution, in time and space, of large-area monitoring efforts of land and water degradation (Shepherd et al. 2015). As monitoring to detect environmental changes requires increasingly fine-scale sample collection, alternative tools for sample analysis that decrease costs and maintain reasonable accuracy may be transformative for surveillance of degradation and restoration.
One promising tool for use in large-area environmental surveillance is visible/near-infrared (VNIR) diffuse reflectance spectroscopy, wherein patterns of electromagnetic radiation absorbed in the visible (350–750 nm) and near-infrared (750–2500 nm) regions are used to predict the physical and chemical characteristics of a substance (soil, plant tissues, sediments) through the use of multivariate statistical models called chemometrics (Shepherd and Walsh 2002). Like all remote sensing, the method depends on the absorbance and reflectance of a particular material at different wavelengths of light depending on its physical (e.g., crystalline structure) and chemical (e.g., mineral composition, organic content) composition. Numerous applications, ranging from food science (Huang et al. 2008), to forage analysis (Norris et al. 1976), and satellite and airborne sensor applications (Kodikara et al. 2012) have demonstrated the practical validity of statistical correlations between the spectral characteristics of materials and a variety of physical and chemical characteristics.
The emergence of lower cost field ruggedized spectroradiometers has helped the technique gain favor for large-area environmental surveillance (Shepherd et al. 2015) and has been successfully applied over a range of terrestrial (Chang et al. 2001; Brown et al. 2006), aquatic (Malley and Williams 1997), and wetland (Cohen et al. 2005) systems as a means to achieve acceptable, and sometimes enhanced and integrative, measurements of soil and sediment properties. Most research has focused on chemical properties of soils. For example, Chang et al. (2001) successfully predicted total C (TC), total N (TN), cation exchange capacity (CEC), moisture content, sand and silt fractions, and extractable Ca in soils throughout the USA, and similar success has been observed in Australia (Dunn et al. 2002; CEC, exchangeable Ca and Mg, and pH in both topsoil and subsoil) and Africa (Shepherd and Walsh 2002; pH, CEC, Ca, Mg, organic C, and soil texture). For a global database of terrestrial soils, Brown et al. (2006) predicted total clay content, clay fractionation (kaolinite vs. montmorillonite), and CEC. Notably, soil nutrients have been predicted across several soil orders (Lee et al. 2003), and even phosphate minerals associated with Fe, Al, Mg, and Ca have been predicted (Bogrekci and Lee 2005a), especially after model corrections for particle size.
Lake Okeechobee is a large (area ~1730 km2) shallow (mean depth ~2.7 m) freshwater lake in South Florida, USA. The lake is highly managed, with a perimeter dike and numerous flow control structures. Water is delivered to the lake from the Kissimmee River and a number of creeks and canals (Havens and Steinman 2015), and from direct rainfall, which accounts for 40 % of water inputs (James et al. 1995). Water is discharged south into the Everglades Agricultural Area and eventually the Everglades, east to the St. Lucie River and west to the Caloosahatchee River (Havens and Steinman 2015). Water quality in the lake is impaired due to excessive nutrient loading (Reddy et al. 2002), and its sediments are a critically important component of phosphorus (P) cycling, since they act as a sink for particulate P and a large internal load of soluble reactive P (SRP; Fisher et al. 2005; Moore et al. 1998). As such, changes in sediment depth, extent, and chemistry are among the most important long-term indicators of ecosystem degradation and recovery and also of the effects of exogenous disturbances (e.g., tropical storms). However, the size of Lake Okeechobee limits sediment sampling density in space and time due to high costs of collecting, processing, and analyzing large numbers of samples.
Though terrestrial soils are different from lake sediments, some properties have been successfully measured with VNIR spectral techniques in lakes (e.g., Ca, Mg, C, and N). Malley and Williams (1997) used spectroscopy to successfully predict concentrations of various heavy metals in sediments of an oligotrophic freshwater lake in Canada. They observed that successful prediction was predicated on associations between metal concentrations and organic matter (OM) content and that OM was the attribute best predicted by near-infrared (NIR) analysis. Similarly, Nilsson et al. (1996) used the reflectance spectra of lake sediments to infer properties of the overlying water column, where NIR-derived models explained 83 % of variability for total P (TP), 68 % for total organic carbon (TOC), and 85 % for pH.
The large littoral marsh in western Lake Okeechobee has substrates more like wetland soils than lake sediment. Cohen et al. (2005) determined that VNIR spectroscopy can be used to predict a variety of chemical characteristics from wetland soils, including P, a key nutrient of interest in Lake Okeechobee’s sediments. As such, there is strong precedent for the efficacy of this approach given a robust calibration library.
Spectral analysis offers significant cost benefits over traditional wet chemistry, in part because multiple sample characteristics can be predicted simultaneously from a material’s spectral profile. For systems like Lake Okeechobee, whose size limits the spatial and temporal density of sampling, using spectral predictions as a component of monitoring efforts could increase the attainable resolution by offering a low-cost alternative to traditional analyses. However, most of the sample costs are incurred in collecting samples, bringing them back to the laboratory, and using extensive preparation (drying, grinding, sieving) before analysis (Nilsson et al. 1996; Malley and Williams 1997). Because field-portable spectroradiometers are now widely available and could be used to collect sediment spectra on site, the possibility exists to scan sediments in the field, obviating the costs associated with sample collection, preservation, transport, and pre-processing. A principal constraint that has limited field application of spectral prediction is that moisture content strongly affects spectra (Kooistra et al. 2003). In settings where moisture contents vary (e.g., terrestrial soils), this may affect model performance sufficiently to render this technique untenable. However, lake sediments are generally saturated, minimizing the effects of variable moisture content. As such, we predicted that wet scanning may allow sufficient performance for lake sediment surveillance. Our specific objectives in this study were to develop a spectral library and associated chemometrics for multiple properties of sediments from Lake Okeechobee and to compare the effectiveness of models derived from spectra of dried sediments to spectra from saturated samples.
Methods
Sample collection and preparation
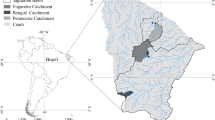
Sample locations spanning Lake Okeechobee were selected based on 174 sites visited during past surveys (Fisher et al. 2005). Eighteen of these previously sampled sites were inaccessible during the dry conditions encountered during our survey, so sediment cores were ultimately obtained at 156 stations (Fig. 1), either using a piston corer (in deep water) or by manually pushing the core tube into the substrate to refusal (in shallow water). Sampling occurred during one field season (summer 2007), with duplicate cores obtained at 12.5 % of sampling sites (one in eight, yielding 19 randomly selected locations). A minimum of 10 cm of site water was left standing above each core, and each was capped and kept on ice during transport to the lab, where it was refrigerated at 4 °C. Cores were extruded inside an N2-purged hood to maintain anaerobic conditions. Surface sediments (0–10 cm) of each core were sectioned for chemical and spectral analysis. Pore water was extracted by centrifugation in airtight containers at 10,000 rpm for 10 min. Syringes were inserted through a rubber stopper in the tube cap to remove pore water without exposing the sample to the atmosphere.

a Map of sampling sites from 2006 sampling effort on Lake Okeechobee (in South Florida, inset). Spatial domains (gray boxes) selected to compare maps derived from measurements with maps from spectral predictions of b Ca, c LOI%, d TP, e TN, and f Mg
Following pore water separation, sediments were dried in an oven at 70 °C for 3 days to determine moisture content and bulk density and then ground in a mortar and pestle and ball-milled to pass through a 20-μm sieve. Part of the sample was retained for spectral analysis, and the remainder was analyzed for a suite of physical and chemical properties according to standard methods: TP (EPA 365.1), TC and TN with a Carlo Erba NA-1500 CNS analyzer, bulk density, HCl-P (EPA 365.1), HCl-extractable Ca, Mg, Fe, and Al (EPA 200.7), KCl-P (EPA 365.1), KCl-NH4-N (Mulvaney 1996), and NaHCO3-P (EPA 365.1). Pore water was analyzed for TP (EPA 365.1), SRP (EPA 365.1), total Kjeldahl N (TKN; EPA 351.2), NH4-N (Mulvaney 1996), pH (EPA 150.1), conductivity (EPA 120.1), as well as Ca, Mg, Fe, and Al (EPA 200.7).
Dried and milled samples were scanned by using a FieldSpec Pro spectroradiometer (Analytical Spectral Devices, Boulder, CO). This device measures diffuse spectral reflectance in the visible (350–750 nm) and near-infrared (750–2500 nm) range in 1-nm bands, by using Spectralon (Labsphere, Hutton, NH) as a white reference. Samples were scanned from below through borosilicate glass dishes (Shepherd et al. 2003). Spectra were resampled at 10-nm increments to reduce data dimensionality prior to statistical analysis. Raw spectra were derivative transformed to highlight spectral response patterns (Fearn 2000), reduce albedo effects, and control for minor differences between batches in light source intensity and effects of differential sample grinding.
To compare spectral prediction accuracy between wet and dry spectra, sediment scans were obtained after dry samples (held dry for 3 months) were rewetted to saturation by using distilled deionized water, and mixed to a homogeneous consistency, then scanned wet after standing for approximately 20 min to ensure saturation and equilibration.
Data analysis
Partial least squares (PLS) regression was used to develop statistical predictions of sediment and pore water chemistry from the spectral data (Nilsson et al. 1996; Dunn et al. 2002; Lee et al. 2003; Bogrekci and Lee 2005b) by using Statistica v. 8.0 (StatSoft Inc., Tulsa, OK). Sites were randomly divided into calibration (67 % of the data) and validation (33 %) sets (Dunn et al. 2002). Separately randomized sets for calibration and validation were used for the wet and dry spectral experiments. This was justified by an ex post facto test on the element with largest prediction differences between dry and wet spectra, Ca, which showed that using the identical sites yielded similar metrics for model performance. Outlier removal and log transformation were used with some analytes (pore water Fe, TP, SRP, NH4-N, KCl-P, and AL and sediment TP, bulk density, and HCl-P) to control for non-normal distributions. Any negative values predicted by the models were assumed to be equal to zero.
Model performance was evaluated by using multiple metrics, including the coefficient of determination (R 2) derived from a linear regression between observed and predicted values, the root mean squared error (RMSE) between predicted and observed values, and the RPD statistic, which is a unitless ratio of the population standard deviation to the RMSE (Chang et al. 2001; Dunn et al. 2002). Model efficiency is inferred from high R 2 and RPD and low RMSE. While no accuracy or usability thresholds exist for R 2 or RMSE, RPDs less than 1.5 are generally considered unsuitable, whereas greater than 2.0 is considered outstanding performance (Chang et al. 2001; Dunn et al. 2002; Cohen et al. 2005); intermediate values are useful in some cases. In addition to evaluating model performance across the suite of analytes, we compared wet and dry model chemometrics as a basis for determining future feasibility of wet scanning.
Map comparisons
Spatial patterns of sediment properties are an important component of monitoring activities in Lake Okeechobee. An open question when using chemometrics to supplement, or even replace, traditional forms of analysis is whether the resulting data are of sufficient quality for mapping purposes. To test the utility of spectral modeling (both wet and dry) for mapping and trend/hotspot detection, separate calibration and validation sets were created for sediment TP, TN, loss on ignition (LOI), and total Ca and Mg. These validation sets were chosen on a spatial rather than random basis and from locations in the lake where spatial features such as gradients or hotspots were observed in maps interpolated from traditional chemical analysis. The areas were of large enough size to encompass roughly one quarter (~40) of the sites. Sample observations outside the target areas were used to calibrate the model.
The map assessment protocol to measure spectral analysis performance in a simulated field setting had four steps. First, select a region for map validation encompassing ca. 25 % (n = 40) of the sites. Second, construct an interpolation surface for that region using an ordinary kriging model using traditional wet chemistry data. We cross-validated these reference maps to estimate and map the spatial interpolation prediction error. This provides a reference level of prediction uncertainty against which we compare the spectral predictions and their associated maps. Third, we used ordinary kriging of spectral predictions over the domain to map chemometric performance; because the calibration data were always outside the area of interest, this map reflects validation performance. Fourth, we compare the first map (wet chemistry measurements) to the second (spectral predictions) to determine the magnitude and pattern of mapping error. All kriging models were run in ArcGIS v 9.3 (ESRI Inc., Redlands, CA) by using 12 lags of ca. 1600 m (except for Ca and TP, which were approximately 650 and 600 m) and modeled semi-variance by using a spherical function applied to the data after second-order trend removal.
Results
Summary statistics were derived for the laboratory measurements of sediment and pore water analytes (Table 1). A wide range of values was observed for all analytes, typically spanning at least two orders of magnitude. Pore water Fe, TP, SRP, NH4-N, and KCl-P and sediment TP, bulk density, HCl-P, and KCl-P were transformed by using natural logs prior to analysis to meet the criteria of normally distributed chemometric model residuals.
Summary statistics for PLS modeling results were tabulated for dry (Table 2) and wet (Table 3) scanning. Most sediment properties were successfully modeled (RPD > 1.5) under both dry and wet conditions. Spectral predictions of Ca, Mg, TC, LOI, bulk density, moisture content, and TN were highly effective (RPD > 2), while predictions for Fe, Al, and TP were moderately effective (1.5 < RPD < 2.0). Sediment SRP was poorly predicted.
RPD scores for sediment predictions from dry sediment scales were, on average, 7 % higher than from wet scans (Fig. 2). Ca and NaHCO3-extractable TP models were markedly better using dry scans, while KCl-extractable P and Mg performed better with wet scans.

Comparison of dry vs. wet scan RPD for sediment spectral results. The dotted line represents the 1:1 line, where wet and dry scans resulted in similar predictive performance
Parallel results for pore water (Tables 4 and 5) indicate that pore water properties were not well predicted by spectral methods, with low coefficients of determination and RPDs below 1.5.
Interpolation maps for observed and predicted values of TP, TN, LOI, Ca, and Mg were developed ((Figs. 3, 4, 5, 6, and 7), and model performance was statistically determined (Table 6). The figures compare map results derived from traditional analytical procedures and results from VNIR prediction (validation performance) of sediment properties at each site. Major analytes of interest for lake management were presented. Spatial regions over which a large chemical gradient or other feature of interest was observed were chosen, in order to compare results from spectral analysis to those obtained from traditional methods.

Maps of observed Ca, predicted Ca, error between predicted and observed, and the standard error of prediction for the same spatial region based on the lake-wide wet chemistry-derived values. All units in g/kg

Maps of observed LOI, predicted LOI, error between predicted and observed, and the standard error of prediction map for the same spatial region based on the lake-wide traditionally derived values. All units in %

Maps of observed TP, predicted TP, error between predicted and observed, and the standard error of prediction map for the same spatial region based on the lake-wide wet chemistry-derived values. All units in mg/kg

Maps of observed TN, predicted TN, error between predicted and observed, and the standard error of prediction map for the same spatial region based on the lake-wide wet chemistry-derived values. All units in g/kg

Maps of observed Mg, predicted Mg, error between predicted and observed, and the standard error of prediction map for the same spatial region based on the lake-wide wet chemistry-derived values. All units in g/kg
The Ca map from laboratory measurements (Fig. 3a) showed a strong gradient from west to east; the attendant spectral prediction map corresponded strongly with this trend (Fig. 3b). The Mg map included two sediment concentration hotspots (Fig. 7a), and the spectral prediction maps captured the spatial location and intensity of these hotspots (Fig. 7b). The TP map included both a strong west to east (Fig. 6b) concentration gradient, as well as a single hotspot (Fig. 5a); the spectral map generally captured this pattern, though a predicted hotspot was present where one was not actually observed (Fig. 5b). The spectral map for LOI also largely predicted regional patterns of elevated organic matter content (Fig. 4b), albeit without the fine detail evident in the map derived from laboratory measurements (Fig. 4a). As with the other analytes, the spectral-derived map for TN (Fig. 6b) compared extremely well with the map using lab measurements (Fig. 6a), though several minor issues with hotspot prediction were evident.
We further evaluated the performance of the spectral prediction by comparing the prediction errors implicit in kriging interpolation (panel c in Figs. 3, 4, 5, 6, and 7) with a map showing the difference between spatial patterns observed using the traditional laboratory methods vs. the spectral predictions (panel d in Figs. 3, 4, 5, 6, and 7). In each case, the intrinsic errors associated with the fitting of a semi-variogram model were larger than the differences between the two sources of mapped data (i.e., traditional and spectral predictions of sediment properties). While both sources of error are relevant, the former can be attenuated with greater spatial sampling intensity, while the latter denote the errors associated with removing wet chemical methods from the monitoring protocol. We interpret the greater magnitude of the kriging errors vis-à-vis the comparative method errors as indicating the considerable benefits of efforts to spatially intensify sampling using VNIR spectral methods. Indeed, the evidence suggests that methodological errors are markedly smaller than interpolation errors, leading to the tentative conclusion that monitoring efforts over large complex lake systems may be better served by increased geographic density enabled by spectral predictions of sediment chemistry than by the greater accuracy but lower spatial resolution afforded by conventional analyses. This may have important implications for designing future monitoring efforts on this and other large, complex lake systems.
Discussion
VNIR model performance
Our results support previous research showing that VNIR spectroscopy can be used as a fast and relatively inexpensive means of predicting the physical and chemical characteristics of Lake Okeechobee’s sediments. Most key sediment properties had statistical prediction efficiencies that suggest either excellent or adequate prediction. Because laboratory analysis represents a significant fraction of the sampling cost, this may present opportunities to significantly augment the spatial and temporal resolution of lake ecosystem monitoring, with consequences for understanding the dynamic nature of these resources (e.g., in response to storms, seasons, and climate variation) and better tracking of degradation and restoration trajectories.
Despite success across a wide variety of analytes, P, the nutrient of major concern to lake managers and the pollutant responsible for eutrophication of the lake (Florida Department of Environmental Protection 2001), was not well predicted. This is notable because previous studies have reported greater success with spectral prediction of P concentrations. For example, Nilsson et al. (1996) were able to derive a model that explained 83 % of the variance in lake water P from spectroscopic analysis of surface sediments. Similarly, Cohen et al. (2005) observed high RPD (2.40) for wetland soil TP, while Dunn et al. (2002) experienced low RPD (1.1) for soil TP in their work. Our TP models, which exhibit RPD values around 1.5, do correspond closely to results of Lee et al. (2003), who saw coefficients of determination (R 2) for TP prediction in the range of 0.52–0.66. This weaker prediction efficiency may be a function of a relatively small calibration set. More likely, however, is that P is present in Lake Okeechobee sediments in multiple forms (various minerals, organic bound), confounding a simple spectral signature approach. Regardless of the origins of weak prediction efficiency, it is clear that spectral predictions of TP are unlikely to supplant traditional laboratory measurements except where spatial scales of heterogeneity are short, necessitating substantially denser spatial sampling to capture extant lake sediment patterns.
We also note that while solid-phase predictions were effective, determination of pore water properties was uniformly poor. Low validation and calibration R 2 values, as well as low RPD scores suggest that VNIR spectroscopy is not suited for predicting pore water chemistry in Lake Okeechobee, at least when scanning is on dry or rehydrated samples (i.e., not fresh samples). This could be due to the transient nature of pore water chemistry, which is susceptible to time-varying environmental factors such as temperature, dissolved oxygen levels, and redox status and not just the composition of the parent substrate matrix in which they reside. However, given the strong expected links between pore water chemistry and the properties of the sediment matrix, it was surprising to see such low prediction performance. We note that associations between laboratory measurements of pore water chemistry and sediment properties were also surprisingly weak. Whether this suggests limitations of the laboratory methods or handling procedures can only be determined with analysis of future samples; if the expected associations between sediment and pore water chemistry emerge in further sampling, it is likely that spectral predictions of pore water chemistry may also improve, perhaps sufficiently to justify use in monitoring applications.
One of the most promising results of our work was the observation that chemometric models developed from saturated samples performed comparably to those from dry samples. This supports what has been a tacit objective of the spectroscopy approach since the outset, namely that the technique obviates the need for sample collection and transport, but rather allows direct field measurements. This promising development suggests that successful field determination of sediment properties is tenable, which could save substantial time and effort and further cut costs associated with sampling. We do note that our wet samples were dried and homogenized samples that had been rehydrated, necessitating further research to determine if accuracy levels can be maintained with wet spectra obtained under field sampling and scanning conditions. However, given that most of the costs for monitoring large and spatially heterogeneous lake ecosystems is embedded in the retrieval and laboratory analysis of samples, even a modest decline in performance under true field conditions may be acceptable given the altered spatial and temporal resolution of monitoring that field scanning enables.
Mapping performance
To explore the efficacy of spectral predictions for capturing the extant spatial patterns in various analytes, we explicitly compared maps made from validation predictions with maps made with the laboratory analyses and quantified the error (i.e., difference between them). In almost every case, the spectrally derived maps compare favorably to those derived from laboratory values, both in the visible spatial gradients and trends and also in specific hotspot detection; for Ca, the maps were nearly identical. This is not entirely surprising for analytes where the prediction utility of the models was judged to be good. However, the concordance between spectral and raw data maps of TP was strong despite lower predictive utility (i.e., low RPD) of chemometrics for TP. This is encouraging and demonstrates that the “application-dependent” proviso for models with intermediate levels of RPD should be taken at face value. That is, analytes for which RPD values are comparatively low should not be written off as lacking spectral prediction utility in all contexts. The utility of VNIR spectroscopy for lake sediment mapping would likely lie in either mapping analyte variation at high spatial resolution (e.g., to better detect boundary locations) because of reduced overall sampling costs. It may also be valuable where the spatial range of variation is small, a condition that makes kriging models with sparse conventional sampling prone to significant map prediction error. Finally, it may demonstrate promise as a large-area surveillance screening tool where larger scale trends in chemical properties can be detected to guide more efficient sampling procedures that employ more precise laboratory methods. We argue that utilizing two tiers of monitoring precision—i.e., lower precision spectral screening tool and higher precision laboratory-based validation—may ultimately provide the appropriate balance for large-area surveillance and monitoring.
One way to evaluate the performance of the spectral predictions for mapping is to consider the difference between the maps from the two analysis methods and the error associated with spatial interpolation. Our rationale is that higher density sampling using spectral methods would lower the spatial interpolation error, but do so utilizing an analytical approach that may sacrifice accuracy. Our findings suggest that the magnitude of spatial interpolation errors is as high or higher than the prediction differences. For example, the RMSE comparing the maps derived from the spectral and laboratory data was lower than the RMSE of the kriging models for all analytes except for TP, which was comparable (RMSE = 239 mg/kg for method comparison vs. 226 mg/kg for spatial interpolation errors). In short, denser spatial sampling enabled using spectral methods would likely benefit spatial characterization of lake sediment properties.
For this spatial analysis comparison, it should be noted that a relatively small dataset was used to calibrate and test the spectral models. One would ideally have as large a calibration set as possible in order to better capture variation in spectral reflectance and other properties. While some point of diminishing returns emerges with adding calibration points as spectral models approach maximum effectiveness, we are confident that our small sample size (105–126 samples, depending on the analyte) presents an important constraint on accurate representation of potential spectral model efficacy. That is, we strongly believe that a larger sample size would yield spectral prediction improvements. A future research priority is to determine the sample library size at which model performance is maximized for Lake Okeechobee’s sediments. Once a suitable spectral library is constructed (based on future field surveys from which both spectral and laboratory analytical approaches are available), it seems likely that spectral monitoring of sediment properties may become cheaper and streamlined.
Considerations for spectral approaches for routine monitoring
The decision to employ VNIR spectroscopy for lake sediment assessment necessarily balances the predictive limitations of spectral methods presented here against savings in both cost and effort, which can be substantial or substantially increased spatial or temporal sampling intensity. Considering only the five analytes mapped using spectral methods, field sampling is the largest cost and would be present regardless of the method used, but processing and analytical costs could be greatly reduced using VNIR in lieu of wet chemistry. In addition to reducing the cost by approximately 25 %, there would also be a significant reduction in time and effort involved in obtaining the data, especially if samples were scanned and discarded in the field. Because one spectrum can be used to determine multiple characteristics of a sample, the savings in cost can increase if spectral methods are used to simultaneously determine multiple analytes of interest.
Conversely, where high accuracy observations are required (e.g., for testing of specific legal criteria or close scrutiny of localized management goals), VNIR technology may not be sufficiently accurate to be a primary analytical tool, and traditional wet chemistry may be preferred. Because of the large size of Lake Okeechobee and the resulting expense and effort associated with high spatial and temporal resolution sampling, the use of VNIR technology could allow for more detailed mapping and monitoring of this system, particularly where a small subset of samples are returned to the laboratory for wet chemistry verification of the spectral predictions. Such a combined approach of conventional sampling with denser spectral in-fill sampling could well yield optimal results. The results from this study contribute to the growing body of evidence that suggests that a move to entirely field-based sampling may be both feasible and desirable for specific environmental monitoring applications and certain analytes of interest. In large-area systems that will be subjected to repeated monitoring and sampling, the reduction in time and cost to collect spectrally derived data offers an opportunity to increase the spatial and temporal resolution of sampling efforts. The ability to obtain real-time chemical predictions from spectra in the field via use of a laptop could also allow for adaptive spatial sampling, allowing managers or researchers to home in on areas of particular interest, where samples might be collected for traditional chemical testing. As the VNIR-associated hardware, software, and statistical analysis methods continue to improve, this resource is likely to be increasingly utilized to increase the efficiency with which monitoring and sampling occur.
References
Bogrekci, I., & Lee, W. S. (2005a). Improving phosphorus sensing by eliminating soil particle size effect in spectral measurement. Transactions of the American Society of Agricultural Engineers, 48(5), 1971–1978.
Bogrekci, I., & Lee, W. S. (2005b). Spectral measurement of common soil phosphates. Transactions of the American Society of Agricultural Engineers, 48(6), 2371–2378.
Brown, D. J., Shepherd, K. D., Walsh, M. G., Mays, M. D., & Reinsch, T. G. (2006). Global soil characterization with VNIR diffuse reflectance spectroscopy. Geoderma, 132(6), 273–290.
Chang, C., Laird, D. A., Mausbach, M. J., & Hurburgh Jr., C. R. (2001). Near-infrared reflectance spectroscopy-principal components regression analyses of soil properties. Soil Science Society of America Journal, 65(2), 480–490.
Cohen, M. J., Prenger, J. P., & DeBusk, W. F. (2005). Visible-near infrared reflectance spectroscopy for rapid, nondestructive assessment of wetland soil quality. Journal of Environmental Quality, 34(4), 1422–1434.
Dunn, B. W., Batten, G. D., Beecher, H. G., & Ciavarella, S. (2002). The potential of near-infrared reflectance spectroscopy for soil analysis—a case study from the riverine plain of South-Eastern Australia. Australian Journal of Experimental Agriculture, 42(5), 607–614.
Fearn, T. (2000). Savitzky-golay filters. NIR News, 6, 14–15.
Fisher, M. M., Reddy, K. R., & James, R. T. (2005). Internal nutrient loads from sediments in a shallow, subtropical lake. Lake Reserv Manag., 21(3), 338–349.
Florida Department of Environmental Protection (2001). Total Maximum Daily Load for Total Phosphorus Lake Okeechobee, Florida.
Havens, K. E., & Steinman, A. D. (2015). Ecological responses of a large shallow Lake (Okeechobee, Florida) to climate change and potential future hydrologic regimes. Environmental Management, 55(4), 763–775.
Huang, H., Yu, H., Xo, H., & Ying, Y. (2008). Near infrared spectroscopy for on/in-line monitoring of quality in foods and beverages: a review. Journal of Food Engineering, 87, 303–313.
James, R. T., Jones, B. L., & Smith, V. H. (1995). Historical trends in the Lake Okeechobee ecosystem II. Nutrient budgets. Arch fur Hydrobiolie, 107, 25–47.
Kodikara GRL, Woldai T, van Ruitenbeek FJA, Kuria Z, van der Meer F, Shepherd KD, van Hummel GJ. 2012. International Journal of Applied Earth Observation and Geoinformation, 14, 22–32.
Kooistra, L., Wanders, J., Epema, G. F., Leuven, R. S. E. W., Wehrens, R., & Buydens, L. M. C. (2003). The potential of field spectroscopy for the assessment of sediment properties in river floodplains. Analytica Chimica Acta, 484(2), 189–200.
Lee, W. S., Sanchez, J. F., Mylavarapu, R. S., & Choe, J. S. (2003). Estimating chemical properties of Florida soils using spectral reflectance. Transactions of the American Society of Agricultural Engineers, 46(5), 1443–1453.
Malley, D. F., & Williams, P. C. (1997). Use of near-infrared reflectance spectroscopy in prediction of heavy metals in freshwater sediment by their association with organic matter. Environmental Science & Technology, 31(12), 3461–3467.
Moore, P. A., Reddy, K. R., & Fisher, M. M. (1998) Phosphorus flux between sediment and overlying water in Lake Okeechobee, Florida: spatial and temporal variations. Journal of Environmental Quality, 27(6), 1428–1439.
Mulvaney R.L. 1996. Nitrogen-inorganic forms. In Methods of soil analysis, part 3—Chemical methods. Madison, WI, USA: Soil Science Society of America.
Nilsson, M. B., Dabakk, E., Korsman, T., & Renberg, I. (1996). Quantifying relationships between near-infrared reflectance spectra of lake sediments and water chemistry. Environmental Science & Technology, 30(8), 2586–2590.
Norris, K. H., Barnes, R. F., Moore, J. E., & Shenk, J. S. (1976). Predicting forage quality by infrared reflectance spectroscopy. Journal of Animal Science, 43(4), 889–897.
Reddy K.R., White J.R., Fisher M.M., Pant H.K., Wang Y., Grace K., Harris W.G. 2002. Potential impacts of sediment dredging on internal phosphorus load in Lake Okeechobee.
Shepherd, K. D., & Walsh, M. G. (2002). Development of reflectance spectral libraries for characterization of soil properties. Soil Science Society of America Journal, 66(3), 988–998.
Shepherd, K. D., Cheryl, A. P., Catherine, N. G., & Bernard, V. (2003). Rapid characterization of organic resource quality for soil and livestock management in tropical agroecosystems using near-infrared spectroscopy. Agronomy Journal, 95(5), 1314–1322.
Shepherd, K. D., Shepherd, G., & Walsh, M. G. (2015). Land health surveillance and response: a framework for evidence-informed land management. Agricultural Systems, 132, 93–106.
Acknowledgments
The South Florida Water Management District provided funding for this research. The authors thank Yu Wang and Gavin Wilson of the Wetland Biogeochemistry Laboratory at the University of Florida for sample analysis assistance and Benjamin Loughran for field assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vogel, W.J., Osborne, T.Z., James, R.T. et al. Spectral prediction of sediment chemistry in Lake Okeechobee, Florida. Environ Monit Assess 188, 594 (2016). https://doi.org/10.1007/s10661-016-5605-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10661-016-5605-8