
Abstract
Septoria tritici blotch (STB) caused by Zymoseptoria tritici is the primary biotic stress for durum wheat production in Tunisia. The present study was aimed to decipher the genetic diversity and population structure of Z. tritici bread wheat isolates; in the northern regions of Tunisia this pathogen infected exclusively bread wheat. A total of 162 single-spore isolates were sampled during the 2015–2016 growing season from nine naturally infected bread wheat fields at four locations in the three main wheat-growing regions in Tunisia, previously reported as hot spots for Septoria disease (Cap Bon, Bizerte, and Beja). Collected isolates were fingerprinted using twelve polymorphic microsatellite markers to assess the genetic diversity and population structure of Z. tritici. All the microsatellite loci were polymorphic and a high genetic diversity was observed within the whole population. The highest Nei’s index value (0.42), Shannon Index (0.84) and private allele numbers (36) were found at the El Haouaria location (Cap Bon region). Furthermore, a moderate genetic differentiation within and among the surveyed locations (73% and 27%) was confirmed by analysis of molecular variance (AMOVA). On the other hand, the STRUCTURE program was shown to be less sensitive in revealing genetic structure especially at low levels of diversity. Thus, information on the genetic structure of the pathogen population collected from bread wheat is useful for designing and implementing durable and effective management strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cereal sector is one of the pillars of Tunisian agriculture in terms of its cultivated area that covers nearly 1.5 million hectares (Ben Hamouda et al., 2016). It is characterized by the predominance of durum wheat (Triticum turgidum L. subsp. durum (Desf.), genome AABB) wich has been cultivated in Tunisia since the Roman era, while bread wheat (Triticum aestivum L. subsp. aestivum, genome AABBDD) was introduced relatively recently after the French colonization (El Felah et al., 2015). Since then, bread wheat was commonly cultivated in a mixture with durum wheat landraces (Ben Hamouda et al., 2016). Tunisian farmers and rural communities used the Arabic word “Gameh” to designate durum wheat against the non-Arabic word “Farina” for bread wheat (Ammar et al., 2011). Tunisia wheat production has often been hampered by low yields due to abiotic and biotic constraints. To compensate for the production deficit, the country has been regularly importing approximately three million tons of wheat every year (Chebil & Frija, 2016).
Septoria tritici blotch (STB), caused by the hemibiotrophic fungus Zymoseptoria tritici (Desm.) Quaedvlieg and Crous (formerly Mycosphaerella graminicola [Fuckel] J. Schröt. in Cohn), is an important disease in North Africa and particularly in Tunisia, where an early, widespread epidemic occurred between 1968 and 1969 (Saari & Wilcoxcon, 1974; Brown et al., 2015). Since the 1990s, epidemics have become recurrent, particularly on durum wheat that is mainly grown in the northwestern regions such as Bizerte, Beja, and Jendouba (Fakhfakh et al., 2011). Nevertheless, Z. tritici also infects the hexaploid bread wheat that is grown exclusively in the northeastern regions of the country, such as in Cap Bon (Bel Hadj Chedli et al., 2018).
Z. tritici has a heterothallic, bipolar mating system (Waalwijk et al., 2002) and undergoes both asexual and sexual sporulation in the field (McDonald & Mundt, 2016). Through asexual reproduction, pycnidiospores formed in the pycnidia, are locally dispersed by rain splash. Pyncidiospores are considered as a secondary source of inoculum, contributing thus to the disease progression during the cropping season (Steinberg, 2015). During sexual reproduction, the pathogen produces fruiting bodies (pseudothecia) that contain ascospores (Eyal et al., 1985) which have the potential to be dispersed by wind and hence constitute the primary inoculum (Ponomarenko et al., 2011; Suffert & Sache, 2011). The sexual cycle of Z. tritici may occur year-round and plays a crucial role in its epidemiology and population structure (McDonald & Linde, 2002; Wittenberg et al., 2009; Nieuwenhuis & James, 2016; Drenth et al., 2019). As a consequence of genetic recombination, most populations of this fungus are highly diverse, even at a very local scale (McDonald, 1997; Zhan et al., 2003; McDonald, 2015).
Many evolutionary events contribute to genetic change within populations. These events include mutation, recombination, gene flow or migration, genetic drift, and selection (McDonald, 1997). In nature, the interaction among these different forces determines the evolutionary trajectory of populations, which is reflected by their genetic structure. According to McDonald and Linde et al. (2002), pathogens that generate the greatest risk of breaking down resistance genes have a mixed reproduction system, a high potential for gene flow, large effective population sizes, and high mutation rates compared to pathogens with strict asexual reproduction that have low potential for gene flow, small effective population sizes, and low mutation rates. Therefore, information on the population structure and the genetic variation of Z. tritici is important for the deployment of efficient control strategies against STB (McDonald & Mundt, 2016).
In many countries, population genetic studies using RFLP, AFLP, and SSR markers, showed that Z. tritici populations are characterized by high genetic diversities and low genetic differentiation at population and country levels (Schneider et al., 2001; Linde et al., 2002; Zhan et al., 2003; Banke & McDonald, 2005). Significant population structure was also reported in many studies, and important genetic differentiation was noted within and among Z. tritici populations sampled at different levels (e.g, region, field, leaf), from several geographical locations including the Czech Republic, Iran, USA and France (Linde et al., 2002; Abrinbana et al., 2010; El Chartouni et al., 2011; Gurung et al., 2011; Drabešová et al., 2013; Siah et al., 2018). In Tunisia, the genetic structure of Z. tritici has been extensively studied, mainly using SSR markers which revealed a high genetic diversity (Berraies et al., 2013; Boukef, 2012; Naouari et al., 2016). However, the majority of studied Z. tritici populations were sampled from durum wheat and to date no significant investigation was conducted to decipher fungal population genetic structure from bread wheat isolates. Such studies may give a better understanding of the epidemiological and evolutionary driving forces; these are particularly important at the El Haouaria location (Cap Bon region), where severe STB outbreaks have occurred recently on the bread wheat landrace ‘Farina Arbi’. The present contribution provides the first description of Tunisian populations of Z. tritici sampled from bread wheat in three northern regions (Beja, Bizerte and Cap Bon) during the 2015–2016 cropping season. This study was part of a larger study that was undertaken to elucidate Z. tritici genetic variation since the recent emergence of STB in a bread wheat field in Tunisia (Bel Hadj Chedli et al., 2018). Population characterization was performed by measuring the patterns of both genetic diversity and population structure of Z. tritici isolates, using 12 microsatellite markers. Here, we address two hypotheses: 1) the existence of high genetic diversity in Z. tritici sampled from bread wheat and, 2) the STB occurrence on the landrace “Farina Arbi” at El Haouaria led to a population structure.
Materials and methods
Fungal sampling and isolation
Bread wheat leaf samples were collected from nine fields in northern Tunisia, where STB infections were reported during the 2015–2016 cropping season (Bel Hadj Chedli et al., 2018) (Fig. 1). The sampled fields covered three regions and four locations: Cap Bon (El Haouaria location), Bizerte (Ichkeul location) and Beja (Goubellat and Oued Zarga locations). Out of the nine assessed fields, four were located in the El Haouaria region where the bread wheat landrace ‘Farina Arbi’ was grown as a monoculture over several years, and two fields were cropped with the variety ‘Salammbô’. Three sampled fields located at Beja and Bizerte were cropped with the varieties ‘Utique’ and ‘Zanzibar’, respectively. Sampling details and GPS coordinates for each field are shown in Table 1.

The geographical locations of sampled Zymoseptoria tritici field populations in northern Tunisia has been shown in different shapes: ✩: Cap Bon región (El Haouaria location), ∆; Bizerte región (Ichkeul location);✧ and ○: Beja region (Oued Zarga and Goubellat, respectively)
Hierarchical sampling was carried out according to McDonald et al. (1999), where infected leaves were obtained from sites approximately 10 m apart within a field. From each field, wheat leaves were randomly collected from different plants and only one isolate was selected from each leaf for genotyping. Mono-conidial isolates were obtained as described by Siah et al. (2010) resulting in 162 Z. tritici isolates (Table 1) that were subsequently grown on PDA medium (potato dextrose agar, 39 g L−1) and stored at −80 °C for further analyses.
DNA extraction and mating type determination
Fungal DNA was extracted using the Sbeadex® mini plant kit (LGC Genomics) performed on a King Fisher KF96 system according to the manufacturer’s instructions. Mating type of each isolate was identified using a multiplex Polymerase Chain Reaction (PCR) amplification of partial mating type loci as described by Waalwijk et al. (2002) (Table 2). PCR reactions for mating type amplifications were performed using a mixture of 2.5 μL (6 μM) of each primer, 5 μL (600 μM) of dNTPs, 5 μL of 10X reaction buffer, 0.25 μL (5 U/ul) of Taq polymerase (Ampli Taq Gold, Roche, Someville MA) and 1 μL (10 to 20 ng/μL) of genomic DNA. PCR reaction volume was adjusted with nuclease-free water to 50 μL per reaction. PCR amplification was done using an MJ Research PT-100 Thermalcycler (Biorad, Hercules, CA) with an initial denaturation at 94 °C for 2 min, followed by 39 cycles of 94 °C for 1 min for denaturation, 68 °C for 30 min for annealing, and 72 °C for 1 min for the extension step, and a final extension at 72 °C for 10 min followed by a 4 °C step for storage before further processing. Amplified products were stained with Gel Red, separated in 1% agarose gels with electrophoresis at 100 V for 45 min and visualized under UV light.
Microsatellite analysis
Twelve pairs of primers corresponding to simple-sequence repeat (SSR) loci representative of the core genome of Z. tritici were used to genotype the 162 isolates. SSR primers were assembled in a single multiplex as described by Gautier et al. (2014). Three isolates were used as references in this study: the two Dutch bread wheat isolates IPO323 and IPO94269 and the Algerian durum wheat isolate IPO95052 (Kema et al., 2018). SSR primers were selected based on length polymorphism criteria (Gautier et al., 2014; Siah et al., 2018), and were amplified using the Type-it Microsatellite kit (Qiagen) following the manufacturer recommendations (Table 2). The forward primers were labelled with four fluorochromes (i.e., Ned, Fam, Pet, Vic) in a way that the same color was given only to markers with a non-overlapping range of allele sizes. Each 25 μL PCR reaction contained 12.5 μL of the Type-it mix, 2.5 μL of primer mix (containing 2 μM of each primer), 2.5 mL of deionized water, 2.5 μL of Q-solution and 5 μL of template DNA (10 ng/μL). PCR reactions were performed with an initial denaturation at 95 °C for 5 min, followed by 35 cycles of 95 °C for 30s, 55 °C for 90s and 72 °C for 30s, with a final extension step of 72 °C for 30 min, using a PT100 Biorad thermalcycler. The PCR products were subsequently run on a 3130 xl instrument (Life Technologies) using the Liz500 size standard. Chromatograms were inspected visually for all markers and individuals using Peak scanner software version 1.0, before final assignment of SSR alleles.
Data analysis
Population subdivision without a priori assumptions was inferred using the STUCTURE 2.3.4 program which uses a Bayesian algorithm to estimate the number of clusters (Pritchard et al., 2000). The admixture model, a burn-in period of 100,000 and 100,000 Markov Chain Monte Carlo iterations were used. The data were analyzed with K ranging from 1 to 10 with 10 repeats for each K. The best estimate of K was based on ∆K as described by Evanno et al. (2005). This approach reduces the risk of overestimating K and provides a correct estimation of the number of clusters. A bar plot for the optimum K was also determined using Clumpak software (Kopelman, 2015). In addition, the relationship between individuals was visualized using a principal coordinate analysis (PCoA) performed with GenAlEx 6.5 software. Furthermore, the population structure analysis was complemented by a weighted neighbor-joining (NJ) tree (with 500 bootstraps), generated using the SSR data, based on the dissimilarity matrix between the individuals, as implemented in the DARwin6 software (Perrier & Jacquemoud-Collet, 2006).
Molecular data analyses were carried out using the GenAlEx software version 6.5 (Peakall & Smouse, 2012). The genetic diversity within locations was assessed by investigating the number of private alleles (alleles found only in a single location), the Nei genetic diversity (H) (Nei, 1973) and the Shannon information Index (I). A Mantel test was also performed using GenAlEx 6.5 to test the correlation between the genetic diversity (GD) and the geographical distance (GGD) between the individuals. Gene flow (Nm) among locations was also determined using GenAlEx 6.5 software. The magnitude of the genetic differentiation between locations was assessed using the Fst fixation index (Weir & Cockerham, 1984), as implemented in GenAlEx 6.5. Genetic variation within and among locations was further partitioned by analysis of molecular variance (AMOVA) under GenAlEx 6.5.
For mating type determination, two approaches were tested. First, the null hypothesis of a 1:1 ratio of two mating types among and within locations, was evaluated using a χ2-test at the significance level of P = 0.05 (Waalwijk et al., 2002). Second, to test the multilocus linkage disequilibrium, the standardized index of association (sIA) was carried out using the LIAN (Linkage Analysis) version 3.7 (Haubold & Hudson, 2000) program. Significant difference from linkage equilibrium was tested using the Monte Carlo random sampling method (1000 iterations), as implemented in LIAN version 3.7.
Results
Population structure and differentiation between locations
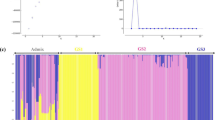
In this study, 162 Z. tritici isolates sampled from bread wheat fields among four locations in northern Tunisia were fingerprinted using microsatellite markers to assess genetic diversity and population structure of the pathogen. Barplot, delta K (ΔK) and L(K) values obtained using STUCTURE output showed a lack of genetic structure among isolates of Z. tritici from the studied locations (Supplementary data Figs. 1, 2 and 3 respectively). No genetic structure as well as no clear geographic origin-based structuring in the Z. tritici population were further confirmed with Clumpak results (Supplementary Data 4). PCoA analysis showed a lack of genetic divergence, where isolates within a location had divergent positions and did not constitute a distinct group (Fig. 2). Furthermore, the hierarchical classification based on the NJ method (Fig. 3) failed to classify isolates to genetic groups with robust bootstrap. Most of the bootstrap values were very low, confirming the lack of genetic similarity . The reference isolates used in this study, the Dutch bread wheat isolates (IPO323 and IPO94269) and the Algerian durum wheat isolate IPO95052, were genetically closely associated with the Z. tritici Tunisian isolates (Fig. 3).

Principal coordinates analysis (PCoA) of Zymoseptoria tritici isolates sampled from bread wheat across four geographic locations in northern Tunisia. Individuals from the same region are marked using the same color used to designate locations in the map (Fig. 1). Dark: El Haouaria population; Red: Bizerte population; Yellow: Oued Zarga population, and Green: Goubellat population. Reference isolates are indicated in Blue

Neighbor-joining tree showing the similarity between the 162 Zymoseptoria tritici isolates sampled from bread wheat across four geographical locations in northern Tunisia. Isolates from the same location are indicated with the same colors used for the PCoA analysis. Numbers above branches represent the percentage of bootstrap values, and values less than 50% are not indicated. The bootstrap values are indicated with the blue color. Dark color is used to designate isolates from the Cap region (El Haouaria location) while the Red color is used to indicate isolates from the Bizerte region (Ichkeul location). Isolates from the Beja region are represented by yellow and green for Oued Zarga and Goubellat, respectively. The reference isolates are indicated with the pink color and circles
Genetic diversity within and among the sampled locations
Out of the 162 Z. tritici isolates genotyped, 128 distinct multilocus genotypes (MLG) were identified (Table 3). At the El Haouaria location, 116 out of the 128 MLG were recorded (Table 3). The Z. tritici population sampled from this location carried the highest number of private alleles (36), whereas populations from Bizerte and Oued Zarga contained one and no private alleles, respectively (Table 3). Of the 12 loci scored, 100% were polymorphic in the El Haouaria location (Cap Bon region) and 75% were polymorphic in the Bizerte and Goubellat locations. Although 3 MLG were detected At the Oued Zargua, the polymorphism was insignificant in this location.. Furthermore, the highest Nei’s gene diversity (0.42) and Shannon’s index (0.84) were recorded in El Haouaria. At the Goubellat and Bizerte locations, Nei’s gene diversities were about 0.35 and 0.37, respectively, while Shannon’s index reached 0.55 and 0.56 respectively (Table 3).
Overall, a significant relationship (P = 0.01) was observed between the geographic distance (GDD) and the Nei’s genetic distance (GD) (Supplementary Data 5) and a moderate level of gene flow (Nm = 1.5) was recorded between locations (Table 3). Furthermore, Fst showed a moderate differentiation (0.23) between the studied locations (Table 3). These results are consistent with the AMOVA analysis, which revealed a moderate but significant level of genetic variation (73%) within and differentiation among (27%) the surveyed locations (P < 0.002, Table 4).
Occurrence of sexual reproduction
One hundred and fourty-two Z. tritici isolates were further assessed for mating types. The two mating types co-existed in the majority of the geographical regions except in the region of Beja (represented by Oued Zarga and Goubellat locations) where a predominance of MAT 1–2 was observed (Table 5). The χ2 test, applied to examine deviation from the null hypothesis of a 1:1 ratio, revealed a non-significant deviation (P < 0.05) from the expected 1:1 ratio in the regions of El Haouaria and Bizerte with frequencies of 40% and 60% for MAT1–1 and MAT 1–2, respectively. A non-significant departure from the 1:1 ratio across the whole bread-wheat Z. tritici population was confirmed by the high multilocus standardized index of association (sIA = 0.16, Table 5).
Discussion
Here, we present the first investigation of genetic variation in populations of Z. tritici sampled from bread wheat across three major wheat-growing areas in Tunisia. However, no genetic structure as well as no clear geographic origin-based structurewas found in the Tunisian Z. tritici population.. The lack of a sharp, genetically distinct clustering was also supported by the results of PCoA analyses that showed an absence of differentiated clusters in the bread-wheat Z. tritici populations sampled from different geographical locations, and confirmed the lack of genetic similarity, i.e., Z. tritici isolates from different regions were grouped together. Neighbour-joining-based cluster analysis did not sharply group the populations according to their geographical origins. The dendrogram showed that isolates from different regions and fields tended to cluster together with the Dutch bread wheat isolates (IPO323, IPO94269) and with the Algerian durum wheat isolate IPO95052 and were even associated with a close genetic similarity. A similar research was conducted by Boukef (2012) using a set of 11 microsatellite loci, where only 4 of them were used in this study (ST9, ST5, ST3C, ST3B), that revealed a lack of genetic structure in the Tunisian durum wheat-derived Z. tritici populations.
The lack of structure has also been reported in another study that looked into the mitochondrial DNA and revealed high levels of gene flow and an active sexual reproduction among Tunisian durum wheat Z. tritici populations (Naouari et al., 2016). Similarity recent research conducted by Mekonnen and collaborators (Mekonnen et al., 2020) did not reveal any population structure in Ethiopian Z. tritici populations. Several earlier studies reported a lack of population structure for Z. tritici at regional and global scales and attributed this result to the long-distance dispersal that probably occurs over a wide geographic area (McDonald et al., 1999; Linde et al., 2002; Zhan et al., 2003). The molecular analyses did not show any apparent population structure of Z. tritici. The absence of genetic structure among isolates of Z. tritici from different localities may be explained by the lack of physical barriers restricting gene flow. The local spatial scales are unlikely to be sites of genetic structure, especially in pathogens with windborn spores (McDonald & Linde, 2002). This result is in contrast to other populational studies, for instance in Iran where the populations of Z. tritici were genetically differentiated between regions, and this genetic differentiation was mostly explained by geographic barriers (e.g. mountains and deserts) as well as differences in climates, cropping systems, wheat cultivars and growing seasons between regions (Abrinbana et al., 2010).
Other possible explanations of this lack of population structure could be related to the sampled area, the population size and the type of marker used (SSR) that cannot accurately capture small genomic variations that would occur at a single-nucleotide level (Väli et al., 2008; Siah et al., 2018). In line with this, Drabešová et al. (2013) reported that different genetic markers revealed different levels of genetic structure for the same sampled population. Recently, using single-nucleotide polymorphisms (SNP), Gibriel (2019) reported a large-scale structural variation in accessory and core chromosomes in the Middle East bread and durum wheat Z. tritici populations.
In this study, the sampling was carried out in the regions of Beja and Bizerte that are considered as hot spots for STB on durum wheat and in the region of Cap Bon, particularly at the El Haouaria location where STB was annually observed on the local bread wheat landrace “Farina Arbi” (Bel Hadj Chedli et al., 2018). This finding could explain the origin of the high levels of genetic diversity observed among Z. tritici isolates sampled from El Haouaria where the highest polymorphism, Shannon’s Index, genetic diversity and private allele numbers were recorded. In contrast, Z. tritici sampled from Beja and Bizerte appeared to have a lower genetic diversity compared to the Cap Bon region, which could be related to the unbalanced sampling size. Interestingly, within the Oued Zarga location, some genetic diversity was revealed by the presence of 3 MLG. In Tunisia, STB is more prevalent on durum wheat and its occurrence on bread wheat was found in a few bread-wheat fields in regions such as Bizerte and Beja (Bel Hadj Chedli et al., 2018), explaining therefore the unbalanced sampling size between these regions. Although our sampled populations may be considered as small and these findings should be taken cautiously.
The high level of genetic diversity observed at El Haouaria is in agreement with previous findings that studied the genetic diversity of this pathogen sampled from natural durum and bread wheat fields across major wheat-producing areas in Tunisia (Naouari et al., 2016; Boukef, 2012) and worldwide (Kabbage et al., 2008; El Chartouni et al., 2011; Gurung et al., 2011; Drabešová et al., 2013; Welch et al., 2017; Siah et al., 2018). The level of genetic variation revealed in this study was supported by the high rate of sexual recombination that could generate new genotypes and increase the genetic variability (Linde et al., 2002; Siah et al., 2010; McDonald & Mundt, 2016). Sexual reproduction in Z. tritici has widely been an effective pathway to increase its fitness to cope with longer-term changes in host resistance or other environmental changes (Kema et al., 2018; McDonald & Linde, 2002; Nieuwenhuis & James, 2016) accelerating pathogen diversification (Zhan et al., 2003; Siah et al., 2018). The predominance of MAT1–2 at Oued Zarga most likely reflected a biased and insufficient sampling of isolates in this region.
Interestingly, AMOVA analysis revealed that 73% of the genetic variation is attributed to differences within locations and 27% to differences among locations. Similar results were observed by Razavi and Hughes (2004) who that indicated that 88% and 12% of the genetic diversity among 90 isolates collected from Saskatchewan (Canada), were related to genetic diversity at intra-population and inter-population levels, respectively. In contrast, using AFLP markers and a total of 221 isolates, Abrinbana et al. (2010) reported the highest Z. tritici genetic variation among wheat regions compared to within regions in Iran. Nonetheless, in our study, moderate differentiation between Z. tritici populations at the regional level was noted by the Fst index, which could be reflected by the moderate gene flow (Nm) between the studied locations. These findings contradict our STRUCTURE results. However, the STRUCTURE program was shown to be less sensitive in revealing genetic structure especially at low level of diversity (Jones & Wang, 2012). In this context, Abrinbana et al. (2010) reported that during the long period of co-evolution of Z. tritici and wheat, gene flow could decrease population differentiation and subdivision. Data analyses using the Mantel test confirmed this result and revealed a significant correlation between the genetic and the geographic distance indicating that isolates close geographically are more similar genetically. In line with this, many researchers reported that genetic differentiation depends on the geographic location studied and the choice of molecular markers (Linde et al., 2002; Abrinbana et al., 2010). In this study, the same MLGs were shared among different populations which could be explained by a high migration level between the different regions or perhaps by the presence of some individuals that had by chance the same multilocus genotype (Gurung et al., 2011). As this fungus has a mixed reproductive system (sexual and asexual), it does not only disperse via clonal pycnidiospores but also via ascospores (sexual spores) that could be considered not only as a primary source of inoculum, but also as a significant source of secondary inoculum strongly involved in wheat plant infection and disease progression (McDonald & Mundt, 2016). Moreover, the sexual recombination is likely the main process responsible for the strong genetic diversity observed in Z. tritici. In this context, McDonald and Linde et al. (2002) reported that when high genotypic diversity occurs among Z. tritici populations sampled from different environments, the major resistance genes may rapidly break down and should be deployed cautiously. To our knowledge, this work is the first study assessing genetic diversity of Z. tritici populations sampled from bread wheat in Tunisia at regional and location scales. This research could provide new information to further explore the epidemiology and management strategies of STB disease on bread wheat in northern Tunisia.
Conclusion
Septoria tritici blotch disease poses a serious threat in Tunisia, mainly on durum wheat. Recently, STB was found to infect bread wheat fields in the North-East of the country particularly, at the El Haouaria location where the local variety “Farina Arbi” was cropped exclusively. Thus, information on the genetic structure of the pathogen population collected from this variety is useful for designing and implementing durable and effective management strategies. This study revealed a high genetic variation, where 73% of the total genetic diversity resides within population and a high likelihood sexual recombination occurs within bread wheat Z. tritici population in northern Tunisia. Among the tested populations, those from El Haouaria displayed the highest genetic diversity, confirming earlier studies that this location is a hot spot for Z. tritici in bread wheat, and also can serve as an excellent location to screen bread wheat germplasm for resistance against STB. Furthermore, a moderate genetic differentiation with a moderate gene flow between the sampled locations, and a lack of population structure for isolates were further demonstrated. Additional samplings from several bread and durum wheat growing areas in Tunisia are needed to confirm this result and to determine the causes of this lack of genetic structure. These investigations could be carried out on populations coming from different years, at different periods during the cropping season and from different host species to gain insight into bread vs. durum wheat-derived populations.
References
Abrinbana, M., Mozafari, J., Shams-Bakhsh, M., & Mehrabi, R. (2010). Genetic structure of Mycosphaerella graminicola populations in Iran. Plant Pathology, 59, 829–838. https://doi.org/10.1111/j.1365-3059.2010.02309.x
Ammar, K., Gharbi, M. S., & Deghais, M. (2011). Wheat in Tunisia. In A. P. A. Bonjean & M. WJ van Ginkel (Eds.), The world wheat book: A history of wheat breeding. Vol. 2 (pp. 443–463). Lavoisier.
Banke, S., & McDonald, B. A. (2005). Migration patterns among global populations of the pathogenic fungus Mycosphaerella graminicola. Molecular Ecology, 14, 1881–1896.
Bel Hadj Chedli, R., Ben M’Barek, S., Yahyaoui, A., Kehel, Z., & Rezgui, S. (2018). Occurrence of Septoria tritici blotch (Zymoseptoria tritici ) disease on durum wheat, triticale, and bread wheat in northern Tunisia. Chiliean Journal of Agricultural Research., 78(4), 559–568. https://doi.org/10.4067/S0718-58392018000400559
Ben Hamouda, H., Ben Youssef, S., Sonia Bedhiaf, S., Mohamed Annabi, M., Zied Draief, Z., & Gharbi, M.S. (2016). Status of agricultural innovations, innovation platforms, and innovations investment. Tunisia. 2015 PARI project country report: Republic of Tunisia. Forum for agricultural research in Africa (FARA), Accra, Ghana.
Berraies, S., Gharbi, M. S., Belzile, F., Yahyaoui, A., Hajlaoui, M. R., Trifi, M., Jean, M., & Rezgui, S. (2013). High genetic diversity of Mycospaherella graminicola (Zymoseptoria tritici) from a single wheat field in Tunisia as revealed by SSR markers. African Journal of Biotechnology., 12, 1344–1349. https://doi.org/10.5897/AJB12.2299
Boukef, S. (2012). Structure des populations, spécificité parasitaire et évaluation de la résistance aux fongicides chez Zymoseptoria tritici, agent de la septoriose du blé en Tunisie. Institut National Agronomique de Tunisie, 1–102.
Brown, J. K. M., Chartrain, L., Lasserre-Zuber, P., & Saintenac, C. (2015). Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genetics and Biology, 79, 33–41. https://doi.org/10.1016/j.fgb.2015.04.017
Chebil, A., & Frija, A. (2016). Impact of improving water-use efficiency on its valuation: The case of irrigated wheat production in Tunisia. African Journal of Agricultural and Resource Economics, 11(2), 131–140.
Drabešová, J., Ryšánek, P., Brunner, P., McDonald, B. A., & Croll, D. (2013). Population genetic structure of Mycosphaerella graminicola and quinone outside inhibitor (QoI) resistance in the Czech Republic. European Journal of Plant Pathology, 135, 135–211. https://doi.org/10.1007/s10658-012-0080-8
Drenth, A., McTaggart, A. R., & Wingfield, B. D. (2019). Fungal clones win the battle, but recombination wins the war. IMA Fungus, 10, 18. https://doi.org/10.1186/s43008-019-0020-8
El Chartouni, L., Tisserant, B., Siah, A., Duyme, F., Leducq, J.-B., Deweer, C., et al. (2011). Genetic diversity and population structure in French populations of Mycosphaerella graminicola. Mycologia, 103, 103–764. https://doi.org/10.3852/10-184
EL Felah, M., Gharbi, M.S., Ben Ghanem, H., & Elloumi, M. (2015). Les céréales en Tunisie entre mythe et réalité. Annales de l’INRAT, 2ème Numéro Spécial Centenaire, 88: 1–17.
Evanno, G., Regnaut, S., & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14, 2611–2620.
Eyal, Z., Scharen, A. L., Huffman, M. D., & Prescott, J. M. (1985). Global insights into virulence frequencies of Mycosphaerella graminicola. Phytopathology, 75, 1456–1462.
Fakhfakh, M. M., Rezgui, S., Jarrahi, T., Zouani, R., Bousalmi, A., Sgatni, K., & Yahyaoui, A. (2011). Mapping distribution of Septoria allowed targeting management options of the disease. In 8th international symposium on Mycosphaerella and Stagonospora diseases of cereals. CIMMYT.
Gautier, A., Marcel, T., Confais, J., Crane, C., Kema, G., Suffert, F., & Walker, A. S. (2014). Development of a rapid multiplex SSR genotyping method to study populations of the fungal plant pathogen Zymoseptoria tritici. BMC Research Note., 7(373), 1,10. https://doi.org/10.1186/1756-0500-7-373
Gibriel, H. (2019). Comparative pathogenomics in the ascomycete plant pathogens Zymoseptoria tritici and Verticillium dahliae. PhD thesis, Wageningen University, Wageningen, Netherlands, 1–232.
Gurung, S., Goodwin, S.B.., Kabbage, M., Bockus, W., and Adhikari, B., T., 2011. Genetic differentiation at microsatellite loci among populations of Mycosphaerella graminicola from California, Indiana, Kansas, and North Dakota. Phytopathology. 101, 1251–1257. https://doi.org/10.1094/PHYTO-08-10-0212.
Haubold, H., & Hudson, R. R. (2000). LIAN 3.0: Detecting linkage disequilibrium in multilocus data. Bioinformatics, 16, 847–848.
Jones, R. O., & Wang, J. (2012). A comparison of four methods for detecting weak genetic structure from marker data. Ecology and Evolution,2(5), 1048–1055. https://doi.org/10.1002/ece3.237.
Kabbage, M., Leslie, J. F., Zeller, K. A., Hulbert, S. H., & Bockus, W. W. (2008). Genetic diversity of Mycosphaerella graminicola, the causal agent of Septoria tritici blotch, in Kansas winter wheat. Journal of Agricultural Food and Environmental Sciences, 2, 1–9.
Kema, G.H.J., Gohari, A.M., Aouini, L., Gibriel, H.A.Y, Ware, S.B., et al. 2018. Stress and sexual reproduction affect the dynamics of the wheat pathogen effector AvrStb6 and strobilurin resistance. Nature Genetics, 50:375–380.
Kopelman, N. M. (2015). CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources, 15, 1179–1191.
Linde, C.C., Zhan J., McDonald, B.A., 2002. Population structure of Mycosphaerella graminicola: From lesions to continents. Phytopathology 92, 946–955. http://doi: https://doi.org/10.1094/PHYTO.2002.92.9.946.
McDonald, B. A. (1997). The population genetics of fungi: Tools and techniques. Phytopathology, 87(4), 448–453. https://doi.org/10.1094/PHYTO.1997.87.4.448
McDonald, B. A. (2015). How can research on pathogen population biology suggest disease management strategies? The example of barley scaled (Rhynchosporium commune). Plant Pathology, 64, 1005–1013. https://doi.org/10.1111/ppa.12415
McDonald, B. A., & Linde, C. C. (2002). The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica, 124, 163–180. https://doi.org/10.1023/A:1015678432355
McDonald, B. A., & Mundt, C. C. (2016). How knowledge of pathogen population biology informs management of septoria tritici blotch. Phytopathology, 106(09), 948–955. https://doi.org/10.1094/PHYTO-03-16-0131-RVW
McDonald, B. A., Zhan, J., & Burdon, J. J. (1999). Genetic structure of Rhynchosporium secalis in Australia. Ecologie and Population Biologie., 89(8), 639–644. https://doi.org/10.1094/PHYTO.1999.89.8.639
Mekonnen, T., Haileselassie, T., Goodwin, S. B., & Tesfayea, K. (2020). Genetic diversity and population structure of Zymoseptoria tritici in Ethiopia as revealed by microsatellite markers. Fungal Genetics and Biology, 141, 305–314. https://doi.org/10.1016/j.fgb.2020.103413
Naouari, M., Siah, A., Elgazzah, M., Reignault, P., & Halama, P. (2016). Mitochondrial DNA-based genetic diversity and population structure of Zymoseptoria tritici in Tunisia. Eurpean Journal of Plant Pathology, 146, 305–314. https://doi.org/10.1007/s10658-016-0915-9
Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the United States of America, 70, 3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Nieuwenhuis, B. P. S., & James, T. Y. (2016). The frequency of sex in fungi. Philosophical Transactions of the Royal Society B: Biological Sciences, 371, 20150540.
Peakall, R., & Smouse, P. E. (2012). GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research-an update. Bioinformatics, 28, 2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Perrier, X., & Jacquemoud-Collet, J.P. (2006). DARwin6 software (http://darwin.cirad.fr).
Ponomarenko, A., Goodwin, S.B., & Kema, G.H.J. (2011). Septoria tritici blotch (STB) of wheat. Plant Heal Instr: 1–7. https://doi.org/10.1094/PHI-I-2011-0407-01.
Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 3, 155–945.
Razavi, M. M., & Hughes, G. R. (2004). Molecular variability of Mycosphaerella graminicola as detected by RAPD markers. Journal of Phytopathology, 152, 543–548.
Saari, E. E., & Wilcoxcon, R. D. (1974). Plant situation of high-yielding dwarf wheats in Asia and Africa. Annual Review of Phytopathology, 12, 49–68. https://doi.org/10.1146/annurev.py.12.090174.000405
Schneider, F., Koch, G., Jung, C., & Verreet, J. A. (2001). Genotypic diversity of the wheat leaf blotch pathogen Mycosphaerella graminicola (anamorph) Septoria tritici in Germany. European journal. Plant Pathology, 107, 285–290.
Siah, A., Tisserant, B., El Chartouni, L., Duyme, F., Deweer, C., Roisin-Fichter, C., Sanssene, J., Durand, R., Reignault, P., & Halama, P. (2010). Mating type idiomorph from French population of the wheat pathogen Mycrosphaerella graminicola: Widespread equal distribution and low but distinct levels of molecular polymorphism. Fungal Biology, 114, 980–990. https://doi.org/10.1016/j.funbio.2010.09.008
Siah, A., Bomble, M., Tisserant, B., Cadalen, T., Holvoet, M., Hilbert, R., Reignault, P., & Halama, P. (2018). Genetic structure of Zymoseptoria tritici in northern France at region, field, plant and leaf layer scales. Phytopathology, 108, 1114–1123. https://doi.org/10.1094/PHYTO-09-17-0322-R
Steinberg, G. (2015). Cell biology of Zymoseptoria tritici. Pathogen cell organization and wheat infection. Fungal Genetics and Biology, 79, 17–23.
Suffert, F., & Sache, I. (2011). Relative importance of different types of inoculum to the establishment of Mycosphaerella graminicola in wheat crops in north-West Europe. Plant Pathology, 60, 878–889. https://doi.org/10.1111/j.1365-3059.2011.02455.x
Väli, Ü., Einarsson, A., Waits, L., & Ellegren, H. (2008). To what extent do microsatellite markers reflect wide-genome genetic diversity in natural populations? Molecular Ecology., 17, 3808–3817. https://doi.org/10.1111/j.1365-294X.2008.03876.x
Waalwijk, C., Mendes, O., Verstappen, E. C. P., de Waard, M. A., & Kema, G. H. J. (2002). Isolation and characterization of the mating type idiomorphs from the wheat septoria leaf blotch fungus Mycosphaerella graminicola. Fungal Genetics and Biology, 35, 277–286. https://doi.org/10.1006/fgbi.2001.1322
Weir, B.S and Cockerham, C.C., 1984. Estimating F-Statistics for the analysis of population structure. Evolution 35: 1358–1370.
Welch, T., Feechan, A., & Kildea, S. (2017). Effect of host resistance on genetic structure of core and accessory chromosomes in Irish Zymoseptoria tritici population. European Journal of Plant Pathology, 150, 139–148. https://doi.org/10.1007/s10658-017-1259-9
Wittenberg, A. H. J., van der Lee, T. A. J., Ben M’Barek, S., Ware, S. B., Goodwin, S. B., Kilian, A., Richard, G. F., Visser, R. G. H., Gert, H. J. K., & Schouten, H. J. (2009). Meiosis drives extraordinary genome plasticity in the haploid fungal plant pathogen Mycosphaerella graminicola. PLoS One, 4(6), 1–10.
Zhan, J., Pettway, R. E., & McDonald, B. A. (2003). The global genetic structure of the wheat pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low mitochondrial diversity, regular recombination, and gene flow. Fungal Genetics and Biology, 38, 286–297. https://doi.org/10.1016/S1087-1845(02)00538-8
Acknowledgments
We kindly acknowledge the “CRP Wheat Septoria Precision Phenotyping Platform, Tunisia” that supported. We thank Dr. Thierry Marcel (INRA-UMR BIOGER, France) for his helpful advice on various technical issues.
Funding
This work was funded by the “CRP Wheat Septoria Precision Phenotyping Platform, Tunisia”.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Chedli, R.B.H., Aouini, L., M’Barek, S.B. et al. Genetic diversity and population structure of Zymoseptoria tritici on bread wheat in Tunisia using SSR markers. Eur J Plant Pathol 163, 429–440 (2022). https://doi.org/10.1007/s10658-022-02486-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-022-02486-x