
Abstract
The highly destructive chestnut blight disease can be successfully controlled by infecting the virulent strain of C. parasitica with the hyperparasitic mycovirus CHV-1. The artificial application of the virus-induced hypovirulence, however, requires that the vegetative compatibility (vc) diversity in the target C. parasitica population be determined beforehand. Conventional vc type determination by pairing the unknown isolate with tester strains of known vc types is time consuming and not always reliable. A genotyping assay was developed that uses DNA from C. parasitica isolates and discriminates the six known di-allelic vic loci and the two mating idiomorphs in two fluorescence-labeled multiplex PCRs. Validation was conducted by successfully genotyping the 74 known European vc tester strains and a conventionally characterized natural population of C. parasitica. Cross-species amplification tests with three Cryphonectria species (C. japonica, C. naterciae, and C. radicalis) revealed a high amplification specificity for C. parasitica, while the analytical sensitivity of the assay was established at 20 pg per reaction of genomic DNA. In conclusion, this fluorescent multiplex genotyping assay offers a simple and high-throughput tool for the characterization of the vegetative compatibility and mating type of C. parasitica at population level, which is particularly important for the application of mycovirus-mediated biological control of chestnut blight.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellular recognition processes play an important role in the sexual and vegetative life cycle of fungi. Sexual reproduction is typically initiated by mate recognition, which—in ascomycete fungi—is controlled by two alleles at a single mating-type locus (Ni et al. 2011). The recognition process between vegetative hyphae is governed by a vegetative incompatibility (vic) system that generally involves several vic loci (Paoletti 2016). Upon fusion of incompatible hyphae, the reaction between incompatible vic gene proteins induces a programmed cell death, thus preventing heterokaryon formation and cytoplasmic exchange. This non-self-recognition process assures the genetic integrity of fungal mycelium and restricts the transmission of deleterious cytoplasmic elements, such as mycoviruses. The fungal vic system has practical relevance if cytoplasmically-transmitted mycoviruses are being used for biological control of fungal pathogens (Ghabrial and Suzuki 2009). Moreover, sexual and vegetative compatibility testing has been widely used to study mode of reproduction and the diversity of fungal populations (e.g. Milgroom and Cortesi 1999; Hoegger et al. 2000; Chang et al. 2014).
Chestnut blight is a highly destructive tree disease involving three trophic levels: the tree host Castanea spp., the fungal pathogen Cryphonectria parasitica (Murr.) Barr., and hyperparasitic mycoviruses, e.g. Cryphonectria hypovirus 1 (CHV-1) (Milgroom and Cortesi 2004; Rigling and Prospero 2018). Cryphonectria parasitica is native to East Asia where it causes minor damage to the local chestnut species, namely, the Chinese chestnut (C. mollissima Blume) and Japanese chestnut (C. crenata Siebold and Zucc.). The pathogen was introduced into North America and Europe, leading to severe disease epidemics on the susceptible American (C. dentata Marsh) and European (C. sativa Mill.) chestnut species (Anagnostakis 1987). Virulent strains of C. parasitica typically induce necrotic lesions (so-called cankers) on the bark of susceptible Castanea species. Perennial cankers are formed on thicker branches or on the trunk, and may enlarge rapidly, girdling the affected tree part. This leads to the death of stems or branches distal to the infection point. Because of chestnut blight, the American chestnut was almost entirely extinct as an important forest tree species in North America. The disease epidemic in Europe took a milder course, and recovery from chestnut blight has been observed in many chestnut growing areas (Heiniger and Rigling 1994). This recovery has been attributed to a viral disease in C. parasitica caused by mycoviruses of the family Hypoviridae, in particular CHV-1, that results in hypovirulence (Choi and Nuss 1992). Cryphonectria parasitica strains infected by CHV-1 exhibit reduced virulence and sporulation capacity compared to isogenic non-infected strains (Peever et al. 2000; Bryner and Rigling 2012). Hypovirulence turned out to be a particularly promising chestnut blight management system because it enables therapeutic treatments of infected chestnut trees (Choi and Nuss 1992; Heiniger and Rigling 1994).
CHV-1 is located in the cytoplasm of C. parasitica, and has no extracellular phase (Nuss 2005). It is transmitted horizontally from an infected to a non-infected fungal mycelium by somatic hyphal fusion (anastomosis) that results in the exchange of cytoplasmic components (Fig. 1). Transmission of CHV-1 via hyphal anastomosis is controlled by a vegetative incompatibility (vic) system (Cortesi et al. 2001) involving several vic genes. To date, six di-allelic vic loci are known which define 26 = 64 vic genotypes or vegetative compatibility (vc) types (Cortesi and Milgroom 1998). The six vic loci have recently been characterized at the molecular level, and studies have revealed complex allelic and non-allelic interactions involving polymorphic and idiomorphic genes (Choi et al. 2012; Zhang et al. 2014). Cryphonectria parasitica strains are vegetatively compatible if they have the same alleles at all vic loci. In this case, anastomoses are readily formed, and CHV-1 is rapidly transmitted between strains. If strains are heteroallelic at one or more vic loci, formation of stable anastomoses is prevented, and virus transmission is restricted (Cortesi et al. 2001). As an exception, one vic locus (vic4) was found not to affect virus transmission (Cortesi et al. 2001). The vic system in C. parasitica has practical consequences for biological control of chestnut blight using hypovirulence. At first, therapeutic canker treatments are only successful if hypovirus transmission is not blocked by vegetative incompatibility. Moreover, high vc type diversity prevents the spread of the hypovirus at the population level, and is thought to be a main reason why hypovirulence could not become established among North American C. parasitica populations (Liu et al. 2000). Therefore, monitoring vc type composition and diversity in C. parasitica populations is crucial for successful biological control using hypovirulence.

Classical determination of the vegetative compatibility in Cryphonectria parasitica. Target isolates are pairwise co-cultured on potato dextrose agar with European tester strains of known vegetative compatibility types. Compatible isolates will merge into a single culture (white arrow), whereas a barrage line is formed along the contact zone between two incompatible isolates (white arrowheads). The hypovirus is transmitted horizontally from an infected to a non-infected fungal individual by somatic hyphal fusion that results in exchange of cytoplasmic components (fusion of two hyphae: black arrow in upper electron micrograph). Pictures: WSL/phytopathology
Since the hypovirus is transmitted into asexual but not into sexual spores, the mode of reproduction of C. parasitica is another important factor affecting the spread of CHV-1 in a fungal population. Furthermore, sexual reproduction may negatively impact CHV-1 transmission by increasing vc type diversity through the genetic recombination of polymorphic vic loci. Cryphonectria parasitica has a single mating type (MAT) locus, which contains either the MAT1–1 or MAT1–2 idiomorph (Marra and Milgroom 2001). According to McGuire et al. (2004), MAT1–1 encodes three genes (MAT1–1-1, MAT1–1-2, and MAT1–1-3) and MAT1–2, one gene (MAT1–2-1). Although C. parasitica seems to be a typical self-incompatible heterothallic ascomycete, in natural populations it is capable of selfing (Marra et al. 2004). Most self-fertile isolates are heterokaryotic for the mating type, i.e., they contain nuclei that are isogenic except at the mating type locus. The progeny of a self-fertile isolate segregates in a 1:1 ratio for mating type (McGuire et al. 2004, 2005). Asexual reproduction results in a clonal population structure, typically characterized by the dominance of a single vc type and mating type (Hoegger et al. 2000; Milgroom et al. 2008). Such clonal populations are particularly suitable for hypovirus invasions (Hoegger et al. 2003).
Despite the importance of accurately identifying vc types of C. parasitica strains for biological control of chestnut blight (Prospero and Rigling 2013), vc type testing is still a demanding task. First, C. parasitica has to be isolated in pure culture from infected bark tissue. Thereafter, isolates need to be co-cultured on a growing medium with European (EU) tester strains of known vegetative compatibility type. Finally, the vc type of the target isolate is determined based on colony morphological features. Compatible isolates will merge into a single culture, whereas a barrage line will be formed at the contact zone between two incompatible isolates (Biella et al. 2002) (Fig. 1). The barrage/merging response is, however, not always easy to assess and, thus, ambiguous vc type assignments are not uncommon. In addition, large series of pairings are needed for populations with high vc type diversity. As an alternative to pairing isolates with tester strains, Short et al. (2015) and Mlinarec et al. (2017) developed PCR assays, which consist of six duplex reactions based on three different cycling protocols. Even though practical improvements have been achieved in this way, the proposed protocols suffer from limitations, such as the high number of individual PCR reactions and gel electrophoreses needed for each fungal individual. Furthermore, the mating type has to be assessed in two additional PCR reactions visualized by gel electrophoresis (Marra and Milgroom 2001).
The present study aims to develop a high-throughput genotyping assay for vic and MAT genes of C. parasitica. Genotyping has a firm place in forensic, diagnostic, and scientific applications, and is mainly performed by PCR with defined primers (e.g., analysis of simple sequence repeats, SSR) or by next generation sequencing techniques (e.g., analysis of single nucleotide polymorphism, SNP). In our specific case, we developed an assay that should be applicable as a multiplex in a 96-well plate format using fluorescent-labeled markers, which can be detected on a capillary DNA analyzer. To simplify procedures, resulting electropherograms should be scored automatically by a size-calling computer program. Our results demonstrate that genotyping vic loci and mating type in multiplex presents significant advantages over the traditional pairing assays or conventional PCR by providing accurate high-throughput results in a short time.
Material and methods
Tester strains and DNA samples
The vic genotypes of the vc types EU-1 to EU-64 at the six currently known vic loci (vic1, vic2, vic3, vic4, vic6, and vic7) were previously determined by Cortesi and Milgroom (1998). Ten additional vc types (EU-65 to EU-74) are officially recognized (Robin et al. 2000; Peters et al. 2014), although, the genetic basis for their incompatibility with the 64 EU vc types remains unknown. All European vc type tester isolates EU-1 to EU-74 are stored frozen at −80 °C in glycerin stocks in the culture collection at WSL.
For primer testing, we selected eight EU tester strains (EU-1, EU-20, EU-33, EU-34, EU-35, EU-42, EU-43, and EU-44) that represent each vic allele and both MAT idiomorphs four-fold. Strains were grown on Potato Dextrose Agar (PDA; 39 gl−1; DifcoTM Laboratories, Detroit, USA) overlaid with a cellophane sheet (BD Difco, Chemie Brunschwig AG, Basel, Switzerland) for a period of 7 d at 25 °C in the dark. Thereafter, mycelia were harvested, transferred to 2 ml Eppendorf tubes, and lyophilized overnight. DNA was extracted using the MoBio PowerPlant DNA Isolation Kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions. The DNA concentration was adjusted equimolar (2.0 ng/μl) for all eight samples. Ten additional DNA samples from our Swiss C. parasitica collection were selected to sequence the vic7 locus, as well as DNA stocks of the C. parasitica population from Claro (n = 58), in Southern Switzerland. The vc type of all isolates from this population was previously determined in pairing assays (Hoegger et al. 2000). All DNA samples were stored at −40 °C in our DNA collection. DNA from stocks was diluted 1:10 in molecular grade water (Merck KGaA, Darmstadt, Germany) on a separate plate before using it for PCR amplifications. All specimen vouchers are listed in Table 1.
Sequence data and primer design
Sequence data from the vic1–vic6 loci (Choi et al. 2012; Zhang et al. 2014), as well as from the MAT locus (Marra and Milgroom 2001; McGuire et al. 2004) were used as the basis for developing our genotyping assay. Two vic7 sequences of the conserved exon region of vic7 allele-1 are deposited in GenBank (KP743739; KP743840; Short et al. 2015), but introns or allele-2 sequences were not available. To ensure a high primer specificity able to distinguish the two vic7 alleles, we amplified both vic7 alleles using primers and protocols developed by Short et al. (2015). Amplicons were then sequenced in both directions at Microsynth AG (Balgach, Switzerland) based on the BigDye Terminator v3.1 cycle sequencing chemistry (Thermo Fisher Scientific corporation, Waltham, Massachusetts, USA).
Primers were designed with the CLC Main Workbench v7 software (QIAGEN) using the following criteria. First, sequences of both alleles per locus were aligned and exons and introns were annotated. Second, regions within conserved exon sequences were targeted to minimize allelic drop-out. The drop-out effect occurs if primer hybridization fails due to sequence polymorphism within the DNA primer-binding sequence, which is more likely to happen in variable intron regions. Third, exon regions with the greatest divergence between both alleles were targeted to increase the primer specificity. Fourth, the amplicon lengths had to differ sufficiently in order to distinguish fluorescence peaks from each another in the electropherograms within the range of 100 bp to 500 bp. All primer sequences were synthesized by Microsynth AG.
Primer testing
For a more cost-effective primer testing, forward primers were initially labeled with the M13-tag 5′-TGTAAAACGACGGCCAGT-3′ according to Schuelke (2000). All PCRs were performed on Veriti Thermal Cyclers (Thermo Fisher Scientific). The optimal annealing temperature was assessed in a temperature gradient increasing by one-degree from 57 to 62 °C under the following conditions: Initial denaturation for 5 min 95 °C; 30 cycles of 30 s 95 °C, 90 s 57–62 °C, and 30 s 72 °C, with a final extension of 10 min 72 °C. The 10 μl PCR mix contained: 1 μl of 2 ng/μl genomic DNA, 200 nM primer per reaction, 3.8 μl water, and 5 μl Type-it Microsatellite PCR kit (QIAGEN). The PCR fragments were verified on 1.5% agarose gels using 1x TBE buffer, 30 min at 80 V/cm for electrophoresis and ethidium bromide (Merck) as the DNA staining dye. From here on, all reactions were set at the optimal temperature of Ta = 60 °C. Testing of the newly designed primer pairs followed Schuelke’s (2000) protocol, but differed in the duplex reactions that contained both primer pairs for one vic locus and, hence, double the quantity of the dye-labeled M13-primer. The PCR mix (10 μl) consisted of 1 μl DNA, each 2.5 μM primer (M13-tailed forward primer: 0.2 μl, reverse primer: 0.8 μl, and dye-labeled M13-primer: 1.6 μl), 0.4 μl water, and 5 μl Type-it. All obtained PCR products were diluted 1:10 in a 1% GeneScan 500 LIZ – Hi-Di formamide mixture (Thermo Fisher Scientific) and sized on the genetic analyzer ABI PRISM 3100 Avant (Thermo Fisher Scientific) using the G5 dye set for fragment analysis.
Primer specificity and sensitivity
The analytical specificity and selectivity of the primer pairs were evaluated (i) in silico using the Primer-BLAST option in the NCBI-suite (https://www.ncbi.nlm.nih.gov), (ii) testing the assay with three closely related Cryphonectria species, i.e., the Asian C. japonica (syn. C. nitschkei, Liu et al. 2003; three isolates), the European C. naterciae (Bragança et al. 2011; two isolates) and C. radicalis (Hoegger et al. 2002; three isolates), and (iii) testing the assay with the 74 EU tester strains and 58 isolates from Claro that were characterized in a former study using the conventional method (Hoegger et al. 2000). All specimen vouchers are listed in Table 1. Analytical sensitivity of the assay was determined using a 10-fold dilution series of C. parasitica DNA from 2.0 ng/μl up to 2.0 E-6 ng/μl concentration. All test assays were performed under the same PCR conditions established in the present study for C. parasitica. Previously genotyped controls and a non-template control consisting of DNAse-free water were introduced within each array.
Results
Primer design and multiplexing
The alleles vic7–1 and vic7–2 were each sequenced successfully from five different isolates from our culture collection. The obtained sequences (GenBank accessions MH836570 – MH836579) were analyzed together with the coding region of vic7–2 of the C. parasitica strain EP155 v2.0 (available from the reference genome portal at the U.S. Department of Energy Joint Genome Institute, JGI; https://jgi.doe.gov) to determine the exon and intron regions in the alignment (Fig. 2). Primers for the vic7 alleles were designed based on this alignment. Previously published sequences were used to design the primer for the other vic loci and for the mating types. Finally, all primer pairs could be placed within exon regions of vic and MAT genes, except Cp-vic7–1-R, which hybridizes for few nucleotides at the 5′ end to an intron region. Cp-vic1a-2 and Cp-MAT1–2 primers were also placed in the exon regions, but each with an intron between them.

Map of partial sequences of six di-allelic vic genes and two MAT idiomorphs of Cryphonectria parasitica with primer binding positions. Sequences are designated with their GenBank accession numbers. Black lines represent introns, while green bars denote exon regions and red bars primer sequences. Black box: Alignment of vic7 sequences. For the vic7 locus, ten sequences were generated newly (MH836570 – MH836579; black lines) and analyzed together with KP743739, KP743740, and jgi|Crypa2|231853| to determine the exon and intron regions in the alignment (green bars)
Since multiplex PCR methods can be prone to amplification failure such as primer dimer interactions, for primer multiplexing, we sorted primers in different combinations of six and eight pairs. Afterwards, we screened for potential primer interactions using the Thermo Fisher Scientific multiple primer analyzer tool (https://www.thermofisher.com). Finally, we sorted all primer pairs into two multiplex assays that showed little interaction in silico and labeled forward primers with fluorescent dyes (cf. Table 2). Since each primer pair exhibits different binding and amplification kinetics, we optimized the DNA amplification efficiency by varying the amount of each primer pair used in multiplex assays. The optimized PCR protocol uses a total volume of 20 μl containing 2.5 μl (multiplex 1) or 1.9 μl (multiplex 2) of genomic DNA (concentration as extracted and subsequently diluted 10-fold), varying primer quantity (Table 2), 10 μl Type-it (QIAGEN) and requires the following conditions: Initial denaturation for 5 min at 95 °C; followed by 25 cycles of 30 s 95 °C, 90 s 60 °C, and 30 s 72 °C; with a final extension of 45 min 72 °C.
Specificity and sensitivity of the genotyping assay
Primer-BLAST in the NCBI-suite compared the primer pairs of the present study with fungal references in GenBank (fungi taxid:4751; April 17, 2018) and revealed that all primer pairs matched all nucleotides for the respective locus of C. parasitica only. The primers for the vic7–2 allele, which was not available in GenBank, were confirmed as not matching any other fungal sequence by Primer-BLAST. All primer pairs were tested on DNA samples from three Cryphonectria species (C. japonica, C. naterciae, C. radicalis). These tests revealed that none of the three species amplified at all possible vic loci, varying from no amplification at all in C. japonica to one amplicon detected in C. naterciae using the Cp-vic3a-1 markers and two weak signals in C. radicalis (vic2 and vic4) (data not shown). The analytical sensitivity test showed that 20 pg/μl initial concentration of input DNA—which equals a concentration of 2.0 pg/ul in the PCR mix—was just at the limit of detection (data not shown).
Allele-specific genotyping
Alleles were automatically sized on GeneMapper 5 using predefined sets of corresponding allele dyes and lengths. The detection threshold was set at 50 height for peak strength by default, i.e., the software cannot identify peaks that fall below this level. For example, analytical sensitivity test showed that a DNA concentration of 20 pg/μl was at the limit of detection for automatic size calling with peak heights of just over the threshold value of 50. Although DNA from the EU tester strains and that from strains from the Claro population was diluted ten-fold before use in the PCR, there were no samples or single loci that exhibited low signal intensity. Thus, each detected peak was displayed in a sizing table directly without lowering the detection threshold. A Microsoft Excel VBA (visual basic for application) macro recoded each allele combination into the corresponding EU and mating type for each isolate (data not shown). All detected vic allele combinations corresponded to the expected vc genotypes for EU-1 to EU-64. Since the vic loci that determine EU-65 to EU-74 are unknown to date, the analysis of these isolates gave the same vic1–vic7 allele combinations as for other EU types (cf. Table 3). Therefore, we verified the (in)compatibility of EU-65 to EU-74 with those EU types, which have the identical allele patterns at vic1a – vic7, by pairing tests according to Table 3. A barrage line developed along the contact zone between all co-cultured isolates indicated the vegetative incompatibility. Although the vic allele combinations have been described previously for all EU tester strains, the corresponding mating types have not. The vic and MAT genotypes for each EU tester strain can be found in Table S1, Supporting Information.
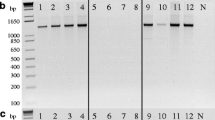
Among the 58 isolates from the natural population from Claro, there was only one discrepancy between the vc type results in Hoegger et al. (2000) and the present study. One isolate identified as EU-6 by Hoegger et al. (2000) was found to be EU-2 in our study. Co-culturing the isolate in question with tester strains of type EU-2 and EU-6 confirmed the EU-2 type of this isolate. Our mating type assay gave the same results as in the previous study, with the exception that we detected both mating types in four samples, i.e., two NED-labeled peaks were present in the same electropherogram (Fig. 3a). To verify this result, we amplified both MAT genes in separate PCRs using the methods described previously by Marra and Milgroom (2001). This confirmed the presence of both mating types in the samples D551, D577, D580, and D590 (Fig. 3b).

Detection of both mating types in the same isolate. (a) Two NED-labeled peaks (black) were present in the same electropherogram of the isolate D577 of Cryphonectria parasitica (orange peaks: DNA size standard). (b) Verification of heterokaryotic strains applying two single PCR reactions according to Marra and Milgroom (2001), which amplify the MAT1–1 (M1) and MAT1–2 (M2) loci specifically, and using the same protocol in a duplex (Dp). The concentration of the PCR product yielded varied considerable and the larger fragment (MAT1–1) from the duplex reaction was in a few cases practically invisible on the gel image
Discussion
Sexual and vegetative incompatibility provide useful genetic markers to study the population biology of fungi (e.g., mode of reproduction, genetic diversity or migration). These markers are also relevant from a practical point of view because both incompatibility systems affect the spread and persistence of biocontrol mycoviruses in populations of pathogenic fungi. The chestnut blight fungus (C. parasitica) and its hyperparasitic mycovirus (CHV-1) represent such a biological control system where sexual and vegetative compatibility testing has been intensively applied (Rigling and Prospero 2018). Conventional characterization of vegetative compatibility in C. parasitica is based on pairing assays involving tester strains of known vc types (Cortesi and Milgroom 1998) or, more recently, on conventional PCR (Short et al. 2015; Mlinarec et al. 2017). Due to the expertise required and difficulties associated with accurately distinguishing the barrage line in pairing assays, false vc type assignments are not uncommon. Additionally, conventional PCR has limitations if a large number of isolates need to be tested. Thus, one of the main challenges at present relates to an accurate molecular technique that becomes indispensable for the analysis of massive sampling. In the present study, genotyping was able to correctly determine each vic genotype of the genetically defined European tester strains EU-1 to EU-64 as well as the previously reported testers EU-65 to EU-74. The genetic basis of these additional EU types has not yet been resolved beyond the known vic loci. Presumably, they differ from EU-1 to EU-64 at additional vic loci. Alternatively, one or several of the known vic loci may have evolved more than two alleles. In any case, once available, new molecular information (additional vic loci or additional alleles) about the incompatibility mechanisms of EU-65 to EU-74 should be included in our assay. Until then, vic genotyping in populations where EU types other than EU-1 to EU-64 have to be expected, should be verified by pairing assays with EU types that share the same vic allele pattern (cf. Table 3).
Careful primer design is critical to ensure amplification of the correct fragment in PCR. In the present study, we used genetically variable regions within exons to develop allele-specific primers in a way that the probability of finding that particular stretch more than once in the genome becomes small. Indeed, in silico primer testing in the NCBI-suite gave no matches with other loci neither in the C. parasitica genome nor in other fungal species. Nevertheless, in vitro, a vic marker (Cp-vic3a-2) amplified in C. naterciae and two in C. radicalis (Cp-vic2–1, and Cp-vic4–2), while no amplification was obtained with the mating type primer for all other Cryphonectria species. These amplifications were unexpected and, even if the amplification yield was low, the obtained fragments showed the expected length for the respective alleles. The present work does not address other species than C. parasitica, nevertheless this finding has important implications for diagnostics: allelic drop-out at most loci must be interpreted as misidentification of the specimen species. Loss of one or more alleles during PCR may be caused by variable mechanisms like single nucleotide polymorphisms in the primer binding sequence. In our specific case, this suggests that species identity requires verification, for instance, by sequencing the barcode locus ITS. Cryphonectria naterciae, for example, has been recovered from symptomatic bark of C. sativa in areas affected by chestnut blight in Portugal (Bragança et al. 2011), indicating that both Cryphonectria species can be present in the same chestnut stand.
Comparative testing of the newly developed genotyping assay with the classical vc and mating typing methods previously applied on the Claro population (Hoegger et al. 2000) showed our assay to have greater sensitivity. First, one isolate was previously classified as EU-6, but our analysis revealed that it clearly belongs to EU-2. These two vc types only differ at the vic4 locus (Cortesi et al. 2007), which is known to induce a weak incompatibility reaction with a faintly marked barrage line formed between co-cultured strains. Thus, false vc type assignments caused by vic4 are not uncommon. Second, the genotyping assay detected both mating types in some isolates. Using state-of-the-art methods at that time, Hoegger et al. (2000) amplified a short, conserved sequence of the HMG box of the MAT1–2 locus and deduced from negative amplification which isolates should belong to the MAT1–1 type. In the present study, we verified the detection of heterokaryotic isolates by applying two single PCR reactions that were developed after Hoegger’s study (Marra and Milgroom 2001). We also tested the same protocol in a duplex reaction that is commonly used in many laboratories. The concentration of the PCR product yielded varied considerably, and the largest fragment (MAT1–1) from the duplex reaction was practically invisible on the gel image in a few cases. Skewed nuclear ratios have been reported in heterokaryotic C. parasitica strains, and may result in underestimating the frequency of MAT heterokaryons in C. parasitica (McGuire et al. 2004). However, our genotyping assay—which uses short fragments for both MAT idiomorphs—was able to detect MAT1–1 accurately, but still at a relatively low concentration level compared to MAT1–2. Detection of both mating types in one isolate is not surprising, and presumably is related to parasexual events (McGuire et al. 2004, 2005) or could indicate the presence of two different strains of C. parasitica.
It has been suggested that vegetative incompatibility can be sidestepped by genome editing C. parasitica strains (Stauder et al. 2019). However, since current European legislation does not allow the release into the environment of genetically modified organisms, such an approach is not applicable in European countries. The fluorescent multiplex genotyping assay presented here is a valuable tool for determining the vegetative compatibility and mating types in C. parasitica populations, which is a prerequisite for the artificial application of CHV-1 to control chestnut blight. In Europe, the mycovirus CHV-1 acts as a successful biological control agent of chestnut blight by reducing the parasitic growth and sporulation capacity of C. parasitica. Individual cankers can be therapeutically treated with hypovirus-infected C. parasitica strains and the hypovirus may subsequently spread to untreated cankers and become established in the C. parasitica population (Rigling and Prospero 2018). As the hypovirus is best transmitted between C. parasitica isolates belonging to the same vc type, the fungal strain carrying CHV-1 should match the vc type(s) present in the target cankers. Ideally, the carrier strain should also have the predominant mating type of the target population, in order to not promote sexual reproduction.
Change history
22 June 2019
Vendor overlooked Fig. 2 & 3 during proof correction and need to be replaced with these.
References
Anagnostakis, S. L. (1987). Chestnut blight: The classical problem of an introduced pathogen. Mycologia, 79, 23–37.
Biella, S., Smith, M. L., Aist, J. R., Cortesi, P., & Milgroom, M. G. (2002). Programmed cell death correlates with virus transmission in a filamentous fungus. Proceedings of the Royal Society of London. Series B: Biological Sciences, 269, 2269–2276.
Bragança, H., Rigling, D., Diogo, E., Capelo, J., Phillips, A., & Tenreiro, R. (2011). Cryphonectria naterciae: A new species in the Cryphonectria–Endothia complex and diagnostic molecular markers based on microsatellite-primed PCR. Fungal Biology, 115, 852–861.
Bryner, S. F., & Rigling, D. (2012). Hypovirus virulence and vegetative incompatibility in populations of the chestnut blight fungus. Phytopathology, 102, 1161–1167.
Chang, S. W., Jo, Y.-K., Chang, T., & Jung, G. (2014). Evidence for genetic similarity of vegetative compatibility groupings in Sclerotinia homoeocarpa. The Plant Pathology Journal, 30, 384–396.
Choi, G. H., & Nuss, D. L. (1992). Hypovirulence of chestnut blight fungus conferred by an infectious viral cDNA. Science, 257, 800–803.
Choi, G. H., Dawe, A. L., Churbanov, A., Smith, M. L., Milgroom, M. G., & Nuss, D. L. (2012). Molecular characterization of vegetative incompatibility genes that restrict hypovirus transmission in the chestnut blight fungus Cryphonectria parasitica. Genetics, 190, 113–127.
Cortesi, P., & Milgroom, M. G. (1998). Genetics of vegetative incompatibility in Cryphonectria parasitica. Applied and Environmental Microbiology, 64, 2988–2994.
Cortesi, P., Mcculloch, C. E., Song, H., Lin, H., & Milgroom, M. G. (2001). Genetic control of horizontal virus transmission in the chestnut blight fungus, Cryphonectria parasitica. Genetics, 159, 107.
Cortesi, P., Rigling, D., & Heiniger, U. (2007). Comparison of vegetative compatibility types in Italian and Swiss subpopulations of Cryphonectria parasitica. European Journal of Forest Pathology, 28, 167–176.
Ghabrial, S. A., & Suzuki, N. (2009). Viruses of plant pathogenic fungi. Annual Review of Phytopathology, 47, 353–384.
Heiniger, U., & Rigling, D. (1994). Biological control of chestnut blight in Europe. Annual Review of Phytopathology, 32, 581–599.
Hoegger, P. J., Rigling, D., Holdenrieder, O., & Heiniger, U. (2000). Genetic structure of newly established populations of Cryphonectria parasitica. Mycological Research, 104, 1108–1116.
Hoegger, P. J., Rigling, D., Holdenrieder, O., & Heiniger, U. (2002). Cryphonectria radicalis: Rediscovery of a lost fungus. Mycologia, 94, 105–115.
Hoegger, P. J., Heiniger, U., Holdenrieder, O., & Rigling, D. (2003). Differential transfer and dissemination of hypovirus and nuclear and mitochondrial genomes of a hypovirus-infected Cryphonectria parasitica strain after introduction into a natural population. Applied and Environmental Microbiology, 69, 3767–3771.
Liu, Y.-C., Durrett, R., & Milgroom, M. G. (2000). A spatially-structured stochastic model to simulate heterogenous transmission of viruses in fungal populations. Ecological Modelling, 127, 291–308.
Liu, Y. C., Linder-Basso, D., Hillman, B. I., Kaneko, S., & Milgroom, M. G. (2003). Evidence for interspecies transmission of viruses in natural populations of filamentous fungi in the genus Cryphonectria. Molecular Ecology, 12, 1619–1628.
Marra, R. E., & Milgroom, M. G. (2001). The mating system of the fungus Cryphonectria parasitica: Selfing and self-incompatibility. Heredity, 86, 134–143.
Marra, R. E., Cortesi, P., Bissegger, M., & Milgroom, M. G. (2004). Mixed mating in natural populations of the chestnut blight fungus, Cryphonectria parasitica. Heredity, 93, 189–195.
Mcguire, I. C., Marra, R. E., & Milgroom, M. G. (2004). Mating-type heterokaryosis and selfing in Cryphonectria parasitica. Fungal Genetics and Biology, 41, 521–533.
Mcguire, I. C., Davis, J. E., Double, M. L., Macdonald, W. L., Rauscher, J. T., Mccawley, S., & Milgroom, M. G. (2005). Heterokaryon formation and parasexual recombination between vegetatively incompatible lineages in a population of the chestnut blight fungus, Cryphonectria parasitica. Molecular Ecology, 14, 3657–3669.
Milgroom, M. G., & Cortesi, P. (1999). Analysis of population structure of the chestnut blight fungus based on vegetative incompatibility genotypes. Proceedings of the National Academy of Sciences, 96, 10518.
Milgroom, M. G., & Cortesi, P. (2004). Biological control of chestnut blight with hypovirulence: A critical analysis. Annual Review of Phytopathology, 42, 311–338.
Milgroom, M. G., Sotirovski, K., Spica, D., Davis, J. E., Brewer, M. T., Milev, M., & Cortesi, P. (2008). Clonal population structure of the chestnut blight fungus in expanding ranges in southeastern Europe. Molecular Ecology, 17, 4446–4458.
Mlinarec, J., Ježić, M., Ćosić, J., & Ćurković-Perica, M. (2017). Multilocus PCR assay reveals high diversity of vegetative compatibility types in populations of Cryphonectria parasitica in Croatia. Plant Pathology, 67, 741–749.
Ni, M., Feretzaki, M., Sun, S., Wang, X., & Heitman, J. (2011). Sex in Fungi. Annual Review of Genetics, 45, 405–430.
Nuss, D. L. (2005). Hypovirulence: Mycoviruses at the fungal–plant interface. Nature Reviews Microbiology, 3, 632–642.
Paoletti, M. (2016). Vegetative incompatibility in fungi: From recognition to cell death, whatever does the trick. Fungal Biology Reviews, 30, 152–162.
Peever, T. L., Liu, Y.-C., Cortesi, P., & Milgroom, M. G. (2000). Variation in tolerance and virulence in the chestnut blight fungus-hypovirus interaction. Applied and Environmental Microbiology, 66, 4863–4869.
Peters, F. S., Bußkamp, J., Prospero, S., Rigling, D., & Metzler, B. (2014). Genetic diversification of the chestnut blight fungus Cryphonectria parasitica and its associated hypovirus in Germany. Fungal Biology, 118, 193–210.
Prospero, S., & Rigling, D. (2013). Chestnut blight. In P. Gonthier & G. Nicolotti (Eds.), Infectious Forest Diseases (pp. 318–339). Wallingford: CABI International.
Rigling, D., & Prospero, S. (2018). Cryphonectria parasitica, the causal agent of chestnut blight: Invasion history, population biology and disease control. Molecular Plant Pathology, 19, 7–20.
Robin, C., Anziani, C., & Cortesi, P. (2000). Relationship between biological control, incidence of hypovirulence, and diversity of vegetative compatibility types of Cryphonectria parasitica in France. Phytopathology, 90, 730–737.
Schuelke, M. (2000). An economic method for the fluorescent labeling of PCR fragments. Nature Biotechnology, 18, 233–234.
Short, D. P. G., Double, M., Nuss, D. L., Stauder, C. M., Macdonald, W., & Kasson, M. T. (2015). Multilocus PCR assays elucidate vegetative incompatibility gene profiles of Cryphonectria parasitica in the United States. Applied and Environmental Microbiology, 81, 5736–5742.
Stauder, C. M., Nuss, D. L., Zhang, D.-X., Double, M. L., Macdonald, W. L., Metheny, A. M., & Kasson, M. T. (2019). Enhanced hypovirus transmission by engineered super donor strains of the chestnut blight fungus, Cryphonectria parasitica, into a natural population of strains exhibiting diverse vegetative compatibility genotypes. Virology, 528, 1–6.
Zhang, D.-X., Spiering, M. J., Dawe, A. L., & Nuss, D. L. (2014). Vegetative incompatibility loci with dedicated roles in allorecognition restrict mycovirus transmission in chestnut blight fungus. Genetics, 197, 701–714.
Acknowledgements
This work was supported by the Swiss Federal Office for the Environment FOEN. The authors thank Paolo Cortesi and Michael Milgroom for sharing vc type tester strains, and two anonymous reviewers for comments on the manuscript. Curtis Gautschi corrected the English text.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
This research does not contain any conflicts of interest, nor research involving humans or animals.
Electronic supplementary material
ESM 1
(PDF 106 kb)
Rights and permissions
About this article

Cite this article
Cornejo, C., Šever, B., Kupper, Q. et al. A multiplexed genotyping assay to determine vegetative incompatibility and mating type in Cryphonectria parasitica. Eur J Plant Pathol 155, 81–91 (2019). https://doi.org/10.1007/s10658-019-01751-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-019-01751-w