
Abstract
The fungal community on the roots, stem bases, stems and grains of organically grown winter wheat was analysed using terminal restriction fragment length polymorphism (T-RFLP) combined with cloning and sequencing of the ITS region. The changes in the composition of fungi in different plant parts and over time as well as interactions between fungi were also investigated. Among 58 fungal taxa found the most common were Davidiella macrospora, Cladosporium spp., Tetracladium maxilliforme, Didymella exitialis, Microdochium nivale and an unidentified species within Ascomycetes. Several potential wheat pathogens were found: Fusarium spp. including F. poae and G. avenacea (F. avenaceum), Microdochium nivale, Oculimacula yallundae, Parastagonospora nodorum and Zymoseptoria tritici and most of them were present on all plant parts. Plant part affected the most the fungal colonization of wheat as was shown both by multivariate analysis of the whole fungal community as well as the analysis based on the identified species. The composition of fungal communities in different parts changed during the growing season but no pattern common for the whole crop could be observed. The most dynamic and significant changes were found among yeasts. Both positive and negative significant interactions between pairwise combinations of pathogens were observed. Positive significant associations were also found between pathogens and other fungi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants are inhabited by fungi that interact with each other and ultimately affect the fitness of natural plant populations and the quality of agricultural crops. Saprophytic fungi living on a plant can protect the plant against pathogens (Liggitt et al. 1997; Perelló et al. 2002). The use of chemical control can, however, change the saprophytic mycoflora and thereby even facilitate the spread of pathogenic fungi (Müllenborn et al. 2008). Fungi colonizing the roots are also very important for plant health and development as they play a role in the suppression of diseases, stimulation of plant growth and in providing nutrients to the plant (Shivanna et al. 1996).
Several reports have described fungal populations on wheat and the most detailed studies including isolation of fungi from roots, culms, leaves, glumes and kernels were performed in Switzerland by Sieber et al. (1988) and recently in Canada by Vujanovic et al. (2012). In Argentina, Larran et al. (2007) isolated endophytic fungi colonizing the above ground parts of healthy wheat plants. Most reports focus however on the fungal microflora of particular parts of the plant such as roots (Bateman and Kwaśna 1999; Lemańczyk and Sadowski 2002), stem bases (Matusinsky et al. 2008), leaves (Blixt et al. 2010; Larran et al. 2002) or grains (Müllenborn et al. 2008; Nicolaisen et al. 2014).
Among the wheat fungal microflora pathogens such as Gaeumannomyces graminis and Rhizoctonia solani can be found on roots (Bockus et al. 2010). Pathogenic Fusarium species, Microdochium nivale, Oculimacula yallundae, Parastagonospora nodorum, Pyrenophora tritici-repentis, Rhizoctonia cerealis and Zymoseptoria tritici are often found on above-ground parts of plants (Bockus et al. 2010). The most serious wheat pathogens in Sweden are Z. tritici, P. tritici-repentis, P. nodorum causing leaf blotch diseases and fungi causing eyespot disease (O. acuformis and O. yallundae) (Wiik 2009). Fusarium species are also common on Swedish wheat (Lindblad et al. 2013) and the incidence of diseases caused by these pathogens is predicted to increase in the future (Roos et al. 2011). During recent years stripe rust (Puccinia striiformis) has also become an increasing problem in Sweden (Hovmøller et al. 2011).
Traditionally, cultivation-dependent methods have been used for the analysis of fungal communities in wheat (Bateman and Kwaśna 1999; Larran et al. 2007; Vujanovic et al. 2012). These methods are time consuming and laborious and unsuitable for analysis of a large number of samples. Moreover, the results may depend on the selected medium and the culture conditions, and therefore do not reflect the original community in the substrate (Pfender and Wootke 1988) since other species may be viable but are not culturable irrespective of medium.
Terminal restriction fragment length polymorphism (T-RFLP) is a high-throughput method used for studying fungal communities. It has been successfully used to analyse the composition of fungal populations in soil (Dickie et al. 2002; Lindahl et al. 2007), wood (Allmer et al. 2006) and wheat leaves (Blixt et al. 2010). T-RFLP allows the identification of individual fungal species in mixed samples by comparison of restriction fragments length from an unknown community (T-RFLP profiles) to a reference database of known T-RFLP patterns (Dickie et al. 2002). The reference database may be derived directly from the analyzed samples through cloning, sequencing and restriction cutting of amplicons (Lindahl et al. 2007).
The overall aim of this study was to investigate the dynamics of fungal populations in organically grown winter wheat by i) identification of fungi inhabiting below- and above-ground plant parts or organs, ii) investigation of changes in the composition of fungal communities during the growing season, and iii) investigation of relationships between pathogens and between pathogens and non-pathogenic fungi colonizing wheat.
This is, to our knowledge, the first published study where the fungal community of the winter wheat’s roots, stem bases, stems and grains was investigated by culture-independent methods simultaneously.
Materials and methods
Sampling and sample processing
Winter wheat plants were collected in 2007 from three organically grown winter wheat fields located in Uppland and Västmanland regions in the eastern central part of Sweden. The previous crop in each field was red clover.
Plants were sampled at growth stages (GS) DC 30–32 (stem elongation, GS1), DC 65–69 (anthesis, GS2) and DC 85–90 (maturity, GS3) according to Zadoks et al. (1974). In each field, one row with 30 sampling sites at 10 m distance was established. On each sampling date, two or more plants were dug up from each site. Depending on the growth stage roots, stem bases, upper stems and grains were analyzed for the presence of fungi (Table 1).
The plants were soaked and washed in running water in order to remove soil. From each of two plants per sampling site (60 plants per field) the main stem was detached and the leaf sheaths removed. The roots were cut off from the stem (just below their points of attachment) and six to eight randomly chosen 3–5 cm root pieces containing root tips were put into a cassette and washed under running tap water over night. Next day the root pieces were washed for 5 min, 5 times, by shaking them in sterile distilled water and after drying put into a freezer (−20 °C).
Stem bases, 3–4 cm long, and upper stems consisting of the highest node and 1-cm segments on each side of it were put into cassettes and washed as described above. Grains were separated from the glumes, ground to a powder in a mill (Retsch, Ninolab AB) and stored in dry conditions at room temperature until processing, which took place within one week.
DNA extraction
DNA was extracted according to the CTAB protocol (Gardes and Bruns 1993) with some modification. Roots, stem bases and upper stems were freeze dried and ground in 2 ml microcentrifuge tubes containing three autoclaved stainless steel nuts with 6.0 mm diameter in a FastPrep® preparation shaker (BIO 101® Systems). The roots were ground once at a speed of 4000 rpm for 40 s. For stem bases and upper stems three 20 s cycles at 4000 rpm were used. For extraction of DNA from grains 150 mg of ground powder were used.
To extract DNA, 1000 μl 3 % CTAB (3 % hexadecyltrimethylammonium bromide, 0.1 M Tris-HCl pH 8.0, 1.4 M NaCl, 0.02 M EDTA) was added to microcentrifuge tubes containing the homogenized plant material. The samples were vortexed and put in a heating-block at 65 °C for 2 h. After 10 min centrifugation at 7000 rpm the supernatant was transferred to a new 1.5 ml tube and 700 μl chloroform: isoamyl alcohol (24:1) was added. The samples were centrifuged for 8 min at 13,000 rpm, the supernatant transferred to a new 1.5 ml tube and precipitated with cold isopropanol. The pellet was washed with 70 % ethanol, re-suspended in 50 μl MiliQ water and stored in −20 °C.
Identification of fungi
For the identification of fungi in plant material, terminal restriction fragment length polymorphism (T-RFLP) was used and a reference database with the T-RFLP patterns of individual fungal species was developed. T-RFLP profiles in the reference database were obtained after cloning and sequencing of the fungal ITS region from 85 samples of roots, stem bases, stems and grains collected during this study. The database was complemented with the profiles of 42 fungal isolates collected from plants from one of the fields in this study, wheat roots from a field with pea as the previous crop, and grains collected from randomly chosen fields and stored in the collection of isolates in the Department of Forest Mycology and Plant Pathology.
In order to identify fungi in plant samples, the ITS region of nuclear rDNA was amplified with the fungal-species specific primer pair ITS1F (Gardes and Bruns 1993) and ITS4 (White et al. 1990) labelled with fluorescent dye D3 and D4 respectively (WellRED, Sigma-Proligo). In 25 μl PCR reaction, a final concentration of 0.2 μM of each primer, 0.2 mM dNTP, 2.75 mM MgCl2, 0.3 U/μl ThermoRed DNA Polymerase (Saveen & Werner AB) and recommended reaction buffer Y was used. The PCR reaction contained 30 ng of total DNA. PCR amplification was performed in 2720 Thermal Cycler (Applied Biosystems) using the conditions: 94 °C for 5 min, 35 cycles of 30s at 94 °C, 30s at 55 °C and 30s at 72 °C, followed by final extension at 72 °C for 7 min. PCR products were separated on 1 % agarose gel stained with ethidium bromide and visualized under UV light (GelDoc, Bio-Rad Laboratories). PCR products were cleaned using JetQuick Kit (GENOMED) according to the manufacturer’s protocol. Purified products (3 μl) were digested in separate reactions with AluI (Roche), CfoI (Promega) and TaqI (Fermentas). Digestions were in a total volume of 6 μl, included 2 U of enzyme, and were incubated for 4 h at 37 °C (AluI and CfoI) or 4 h at 65 °C (TaqI). Restriction digests were purified using Sephadex G-50 Medium (GE Healthcare Bio-science). The length of the terminal fragments was analysed in a Beckman Coulter CEQ™ 8000 Genetic Analysis System using CEQ™ DNA Size Standard Kit-600.
T-RFLP patterns of samples were compared to the reference database using the software TRAMPR (TRFLP analysis and matching package for R) (FitzJohn and Dickie 2007). Fungal taxa were identified if minimum four out of six of the enzyme/primer combinations were present in the sample. The tolerance for the mismatches in peak location was set to 1.5 bases.
To set up the reference database the ITS region from 85 samples of different plant parts were amplified with nonfluorescent primers under the same conditions as described above. PCR products were pooled into seventeen cloning reactions, cloned into the PCR® 2.1- TOPO vector according to the protocol of TOPO TA Cloning Kit (Invitrogen Life Technologies) and transformed into One Shot TOP10 Chemically Competent Escherichia coli. All clones chosen for sequencing were tested for the presence of an insert by PCR with M13 forward and reverse primers. PCR amplicons from cloning were purified with the Agentcourt® AMPure® Protocol 00060IV024. In total, 386 positive clones were sequenced from both ends using ITS1F and ITS4 primers (sequencing by Macrogen, Seoul, S. Korea).
Sequences were manually edited and aligned using the SeqMan II software (DNA Star, Madison, WI, USA). Consensus ITS sequences were matched with DNA sequences from GenBank at NCBI using the BLAST search function (Altschul et al. 1997). The criterion for species identification was minimum 98 % similarity with a known ITS sequence. Sequences were submitted to GenBank under accession numbers: KC694147-KC694161, KC753399-KC753447, KC989057-KC989105.
In order to obtain T-RFLP profiles of the identified fungal species, the representative clones were subjected to T-RFLP analysis as described above for samples.
The isolates included in the reference database were identified by sequencing of the ITS region and subjected to T-RFLP analysis, as described above.
Statistical analysis
Analysis based on the identified species
In this analysis only samples in which the fungi were identified by matching of the restriction profile with the patterns from the reference database were included (N = 610).
Species richness and the composition of fungal communities were analysed in relation to plant part and growth stage. Species richness was analysed in R by calculating species accumulation curves (SAC), showing the relationship between the number of observed taxa and the sampling intensity (Colwell and Coddington 1994). Fungal community composition was compared by calculating qualitative Sorensen similarity indices (CS) (Magurran 1988).
The relationships between the composition of community and environmental variables of plant part, growth stage and field were analysed with the canonical correspondence analysis (CCA) in CANOCO 4.5 (Biometrics-Plant Research International, Wageningen, The Netherlands). The species matrix containing presence/absence data was used in the multivariate analysis. The percentage of the total inertia that was explained by the environmental variables was calculated by dividing the sum of all canonical eigenvalues by the sum of all eigenvalues. The statistical significance of the CCA was evaluated using a Monte Carlo permutation test with 499 permutations.
A series of general linear models were used to examine variation in the occurrence of the various fungal taxa in different plant parts and over time. Logistic regression analysis was used to determine whether the occurrence of the fungal taxa, expressed as the odds of finding a fungus, varied in the four different plant parts (roots, stem bases, stems, and grain). For these analyses, only fungi that occurred in more than 8 samples total were included. This was the minimum number of samples needed for the analysis. Logistic regression was also used for stratified analyses of three plant parts (roots, stem bases, and stems) to see if the occurrence of the different taxa, calculated as the odds, changed significantly during the growing season. The analysis was possible to perform only on the most common taxa, i.e. those that were identified in more than 5 % of all samples.
A log-linear model (Xu et al. 2005) was used to test the significance of co-occurrence of the most common fungi, including plant pathogenic fungi, and these analyses were also stratified by plant part. The general linear models analyses were carried out using the glm function in the R version 2.8.1. Upper and lower 95 % confidence intervals were calculated where relevant.
Analysis of the whole fungal community
For the analysis of the whole fungal community T-RFLP data from 790 samples were uploaded to the online TRFLP analysis software package T-REX (http://trex.biohpc.org/) (Culman et al. 2009). True peaks were identified for all samples of which the area exceeded the standard deviation (assuming zero mean) computed over all peaks and multiplied by one (Abdo et al. 2006). The peaks were then aligned using 1.5 bp clustering threshold in T-REX.
An Additive Main Effects Multiplicative Interaction (AMMI) model within T-REX was used to analyse differences in the fungal communities between plant parts, growth stages and fields (Culman et al. 2009). Binary data (presence/absence of peaks) rather than peak height or peak area were used in the analysis since this data type is the most informative and also recommended for multivariate analysis (Culman et al. 2008).
The AMMI model uses analysis of variance (ANOVA) to first divide the variation into main effects and interactions, and then applies PCA to the interactions to create interaction principal components axes (IPCAs) (Gauch 1992). The variation from the interactions between the main effects (here T-RFs vs. environments) reflect how microbial community profiles (T-RFs) differentially respond to the environments (Culman et al. 2008) and was therefore the most interesting for our study. The analysis was done using the combined data generated by TRFLP of all three different digestion enzymes. Scatterplots of the first two axes of AMMI were graphed with Minitab® Statistical Software v.16 (State Collage, PA).
Results
Reference database and identification of fungi in plant material
T-RFLP patterns of 67 fungal taxa occurring on wheat were collected in the reference database. Among them, 25 fungal taxa were identified by cloning and sequencing of the ITS region from plant samples and 21 were obtained by sequencing of fungi isolated from different parts of the plants. Twenty-one fungal taxa included in the database were identified both as clones and isolates.
A comparison of fungal sequences to reference sequences at NCBI resulted in 46 and 15 sequences identified to species and genus level respectively. Four sequences were identified to order and 2 sequences represented unidentified Ascomycetes.
For most of the fungal taxa a unique restriction pattern was obtained. For fungi from the genera Fusarium and Cladosporium the cutting of the ITS region with restriction enzymes Alu I, Cfo I, Taq I gave fragments very similar in length (Online Resource 1). This means that the species Fusarium culmorum, F. graminearum and F. sporotrichioides as well as Cladosporium cladosporioides and C. tenuissimum could not be distinguished during fragment analysis (TRAMPR). In the analysis of fungal communities these species are therefore presented as Fusarium spp. and Cladosporium spp.
Identification of fungi in the samples was performed by comparing the restriction patterns obtained from the samples to the fungal profiles collected in the reference database. Out of 810 samples collected, 790 samples were analysed by T-RFLP. Twenty samples were excluded from the fragment analysis due to lack of a high quality PCR product after amplification of the ITS region. Matching of T-RFLP profiles of samples with fungal profiles from the reference database identified fungi in 610 samples. These samples were used in analyses based on identified species. The identification of fungi failed for 180 samples.
Diversity of fungi
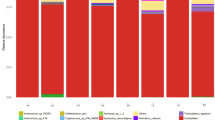
A total of 58 fungal taxa were identified using T-RFLP analysis (Fig. 1). Although none of the accumulation curves of fungal taxa reached an asymptotic value a decreased rate of accumulation of new taxa was observed with increasing sampling effort (Fig. 2).

Fungal taxa identified in different parts of winter wheat from three organic fields during one growing season using T-RFLP. The graph shows the number of samples in which the fungus was detected. Total number of samples N = 610, and for each plant part: roots N = 194, stem base N = 194, stem N = 141, grains N = 81. The taxon code used in Fig. 3 is given in parenthesis. Cladosporium spp. represents Cladosporium cladosporioides and Cladosporium tenuissimum. Fusarium spp. represents Fusarium culmorum, Fusarium graminearum and Fusarium sporotrichioides. ns not significant in logistic regression analysis, nt not tested

Richness of fungal taxa detected in different parts of winter wheat, as expressed by the relationship between cumulative number of taxa found and sampling intensity
Six fungal taxa from the reference library: Gaeumannomyces graminis, Mortierellales, Mucor sp., Pleosporales, Rhizoctonia sp. and Sydowia polyspora were not detected in the plant material.
The majority of fungi colonizing the winter wheat belonged to the phylum Ascomycota (75.8 %). The most common species were Davidiella macrospora, Cladosporium spp., Tetracladium maxilliforme, Didymella exitialis, Microdochium nivale and an unidentified ascomycete. These were all found in more than 20 % of the samples. One fifth of the identified fungal taxa belonged to Basidiomycota, and out of them the yeasts Cryptococcus victoriae and Cryptococcus tephrensis were the most frequent; 16.6 % (found in 101 samples) and 13.6 % (found in 83 samples) respectively. The subphylum Mortierellomycotina was represented by one genus, Mortierella, which was found only in roots. Since the ITS primers that were used do not work for identification of rust fungi and oomycetes the presence of species from those groups in the samples is not known. Among the fungi colonizing wheat, six potential wheat pathogens identified to species were found: Fusarium poae (found in 25 samples - 4.1 %), Gibberella avenacea (found in 60 samples - 9.8 %), Microdochium nivale (found in 132 samples - 21.6 %), Oculimacula yallundae (found in 106 samples - 17.4 %), Parastagonospora nodorum (found in 101 samples - 16.6 %) and Zymoseptoria tritici (found in 52 samples - 8.5 %). Other pathogens from the genus Fusarium were found but because of similar TRFLP profiles they could not be identified as separate species.
The highest number of taxa was found in roots (46 taxa), followed by stem bases (42), stems (36) and grain (29) (Fig 2). The average number of taxa per sample was 4.5 for roots (range 1–17), 2.6 for stem bases (range 1–16), 3.9 for stems (range 1–13) and 5.1 for grain samples (range 1–11).
Comparison of the fungal communities on different wheat parts using the Sorenson similarity indices (Cs) showed moderate to high similarities in community composition. The composition of the fungal population of stem bases had the highest similarity to the communities found on roots and stems (Cs = 0.75), whereas the biggest differences were observed between the populations on grains and roots (Cs = 0.62). Out of 46 fungal taxa identified on roots 23 were not detected on the grains. The most common among them were Tetracladium maxilliforme, an unidentified ascomycete, Myrmecridium schulzeri and Cyathicula sp., identified in 64.9 %, 47.9 %, 23.2 % and 23.7 % of the root samples respectively (Fig. 1). Among fungi colonizing the grains, six taxa were found there but not on roots (Fig. 1). Most of the fungi colonizing grains also occurred on stem bases and stems. 12 taxa were specific only for one plant part. Chaetomium globosum, Fusarium redolens, Ilyonectria destruktans, Lachnum sp., Nectria sp., Cyathus olla and Mortierella sp. were unique for roots. Podospora sp., Sarocladium strictum and Tubaria prestans were found only in stem bases. Penicilium sp. was unique for stems and Pyrenochaeta sp. was unique for grains (Fig. 1). Seventeen taxa including four potential pathogenic species and the genus Fusarium, were found on all plant parts. The exceptions among the pathogens were Z. tritici and O. yallundae, which were not detected in grains.
Although pathogenic fungi were found in all plant parts differences between organs colonized were observed (Fig. 1). The most frequently detected pathogens in roots were M. nivale, G. avenacea and O. yallundae which were found in almost 20 %, 18 % and 15 % of the samples, respectively. These pathogens dominated also on stem bases where O. yallundae and M. nivale occurred in more than 30 % of the analyzed stem bases (P < 0.001). On stems the most frequently observed pathogen was Z. tritici, found in 27 % of the samples (P < 0.001), whereas on grains P. nodorum and F. poae occurred with the highest frequencies; found in 47 % and 25 % of the samples, respectively (P < 0.001).
Significant differences in the distribution of fungi between plant parts were observed for 35 fungal taxa (Fig. 1).
In the CCA model the relationship between identified fungal species and environmental variables was tested. All analysed variables, i.e. plant part, growth stage and field, were found to have small but significant effects on the composition of fungi in wheat (Table 2, Fig. 3). The most important source of variation was plant part, which explained 8.2 % of the total variance in fungal species. Growth stage and field explained 2.0 % and 1.5 % of the total variance in fungal community, respectively.

Seasonal and plant part dependent variation of fungal community analysed by canonical correspondence analysis. R: roots, SB: stem base, S: stem, G: grain, GS1: growth stage1 (stem elongation); GS2: growth stage 2 (anthesis), GS3: growth stage 3 (maturity); taxon code is explained in Fig. 1
The AMMI analysis of the whole fungal population also showed that fungal community composition was the most affected by plant part as the samples grouped into four distinct clusters corresponding to roots, stem base, stem and grains (Fig. 4). Clusters of roots, stem base and stem were located close to each other.

AMMI analysis of fungal T-RFLP data from roots (R), stem bases (SB), stems (S) and grains (G) of winter wheat during one growing season
Analysis of the source of variation showed that plant part had the greatest variation of interaction effects (36.2 %), whereas the variation of interaction effects for growth stage and field was lower and stood at 10.19 % and 7.66 %, respectively (Table 3).
Seasonal variations
The fungal community colonizing roots and above ground parts of wheat changed during the growing season (Fig. 5, Online Resource 2). It is shown by the odds of finding fungi in plant parts obtained as a result of logistic regression analysis. The odds increased, decreased or fluctuated depending on fungus and plant part. As an example: the odds of finding the pathogen Gibberella avenacea in roots decreased from 0.25 in young plants to 0.06 in mature plants, whereas in the stem bases the odds increased from 0.02 to 0.11 during the growing season. The changes observed for this fungus in roots were statistically significant.

The occurrence of the most common fungal taxa on winter wheat during one growing season analysed by logistic regression and shown as the odds of finding fungi in plant parts. Statistically significant changes are marked with different letters. *growth stage 1, 2, 3: GS1 (stem elongation), DC30–32; GS2 (anthesis), DC 65–69; GS3 (maturity), DC 85–90 according to Zadoks et al. (1974)
In roots of young plants D. exitialis, one unidentified ascomycete species and two pathogens, G. avenacea and M. nivale, were detected more often than in roots of mature wheat plants (confidence limits not overlapping) (Fig. 5, Online Resource 2). The opposite situation, but not statistically significant, was observed for M. schulzeri and the pathogenic fungus O. yallundae.
Three Cryptococcus spp. were detected with high frequency in stem bases early in the growing season (May) but they were absent in this plant part in the end of the season (July/August). In mature plants C. victoriae and C. tephrensis were frequently found on higher parts of the stem. A similar variation was observed for the pathogen Z. tritici, which was detected more often in stem bases of young plants than in older plants. The changes observed for this fungus in stem bases were statistically significant. In mature plants this fungus was not found in the stem bases but on higher parts of the stem.
Between anthesis and maturity of the wheat there was a significant decrease in the detection of yeasts, Sporobolomyces roseus and Udeniomyces pannonicus, in stems while the detection of pathogens, M. nivale and P. nodorum, in stems increased significantly during this period.
Although the fungal community colonizing the roots changed over time, the Sorenson similarity index was stable (Cs = 0.78–0.81). For stem bases it fluctuated over time, showing the highest turnover between fungi inhabiting young and mature plants (Cs = 0.52).
Relationships among fungi
Among 63 pair-wise combinations of common wheat pathogens detected in roots, stem bases and stems only 10 showed significant interactions (Table 4). Most of them were positive, which means that the presence of one pathogen favoured the presence of the other. The most significant interactions were observed in the upper part of the stem whereas in the roots only one interaction out of 21 tested was significant.
Fusarium spp. showed the most interactions with other pathogens. In the stem there was a positive interaction between Fusarium spp. and F. poae and G. avenacea. At the same time Fusarium spp. showed negative interaction with P. nodorum and Z. tritici. In the stem base positive interaction was observed between Fusarium spp. and Z. tritici while there was a negative interaction between M. nivale and O. yallundae.
The strongest positive interactions (P = 0.001) were observed in three combinations of pathogens: G. avenacea – M. nivale in roots, P. nodorum – G. avenacea in stem bases and P. nodorum – Z. tritici in the stem.
Almost all pathogens, except for F. poae, showed significant interactions with the most common saprophytic fungi and/or yeasts (Table 5). Most of the interactions were positive. The only negative interaction was observed between M. nivale and D. macrospora in grains.
The pathogen P. nodorum was the fungus which showed the most positive interactions with the common non-pathogenic fungi. Positive interactions were detected with yeasts in the stem and with the saprophytic fungi Cladosporium spp. and D. macrospora in all plant parts except for the roots. In the stem Z. tritici also showed positive interactions with these fungi. In roots positive interactions were observed between G. avenacea and all non-pathogens tested except for Cladosporium spp. M. nivale also showed positive interactions with all saprophytic fungi in the roots.
A formal disease assessment was not carried out in any of the three fields, but a general overview showed no remarkable occurrence of any specific disease on any part of the plants. Disease surveys in 2007 from the same area as where the fields were located report weak attacks of eyespot (Oculimacula yallundae) and tan spot (Pyrenophora tritici-repentis), and few cases of take-all (Gaeumannomyces graminis). Septoria tritici blotch (Zymoseptoria tritici) was the most common leaf spot disease but it did not exceed 20 % incidence until after the end of anthesis to early milk stage (DC 69–73) (Waern and Sandström 2007).
Discussion
The fungal community colonizing organically growing wheat represents fungi of different ecology. They can be classified according to their functions as wheat pathogens (incl. minor pathogens), grass pathogens, saprophytic filamentous fungi often present on wheat, yeasts, and fungi with undetermined function in this system.
The most important factor affecting the colonization of wheat by fungi was plant part as was shown both by the multivariate analysis based on individual species as well as on the whole fungal population. Both methods showed also that differences in fungal communities between plant parts were greater than between growth stages or fields. Organ specificity has been reported in several studies (Larran et al. 2007; Sieber et al. 1988; Vujanovic et al. 2012) indicating that fungi could adapt to the particular microecological and physiological conditions present in a given organ. Species richness and diversity was the highest in roots. It could be attributed to the special conditions in this plant part. Roots create a relatively stable and nutrient rich environment, compared to above ground organs, which favours the survival of many fungal species (Vujanovic et al. 2012). Although the fungal community of roots was the most diverse it was closely related to the community on stem base and stem, as was shown by the multivariate analysis.
Most fungi were able to colonize more than one plant part. Four dominating species D. macrospora, Cladosporium spp., D. exitialis and M. nivale were found both on roots and above parts of wheat which indicates that these fungi were the best adapted to colonize wheat.
Seasonal variation in the composition of fungal communities in different parts of wheat was observed during this study. In roots the contribution of non-pathogenic fungi decreased during the growing season, while the contribution of pathogens varied depending on the species. Some pathogens, such as M. nivale and G. avenacea, causing pink snow mold and Fusarium Head Blight, were more common in young roots, whereas O. yallundae, causing eyespot, was more common in old roots. Similar patterns of colonization of wheat roots by saprophytic fungi have been reported by Lemańczyk and Sadowski (2002). They also observed that the pathogens became dominant with the development of the plant.
In stem bases M. nivale and O. yallundae, but not G. avenacea, were more common in young plants. The colonization of different parts of wheat by the pathogens depends on their aggressiveness as well as on the tissue specificity or systemic spread from the point of infection. During the growing season more aggressive fungi, like O. yallundae, could infect the plant and replace the weaker ones. Matusinsky et al. (2008) observed that Oculimacula spp. and R. cerealis infections increased with the development of the crop but these pathogens infected only plants previously colonized by M. nivale. Decrease of non-pathogenic fungi may also result from rapid colonization of tissue by more aggressive fungi in which case the non-pathogen would not be able to function as an effective antagonist against the pathogen.
On the above ground parts of wheat the most dynamic and significant changes were observed among yeasts. Yeasts are commonly found both on healthy wheat leaves and stems (Larran et al. 2002) as well as on leaves infected by pathogens (Blixt et al. 2010; Perelló et al. 2002). Our results show that yeasts also colonize roots, stem bases and grains. A clear decrease of white-pigmented yeasts from the genus Cryptococcus observed in stem bases late in the season can be due to the strong competition for nutrients with pathogens and saprophytes colonizing stem bases during plant development. Environmental conditions could also influence the growth of yeast in stem bases. On the upper parts of the stem the pattern of colonization was different. Two species of Cryptococcus were abundant in the uppermost node of the mature plants and even during anthesis, while Udeniomyces pannonicus and Sporobolomyces roseus occurred in high abundance in the stem during anthesis and then decreased significantly.
Some pathogens were found on plant parts where they normally do not occur. Examples are the leaf pathogens Zymoseptoria tritici, causing Septoria tritici blotch, and Parastagonospora nodorum, causing Stagonospora nodorum blotch, which were found on stem bases and sporadically on roots, and the stem base pathogen Oculimacula yallundae found on the stem around the uppermost node. These infestations could come from surviving inoculum on old crop debris or perennial grass weeds or from airborne ascospores. In organically grown wheat seedborne P. nodorum could be another inoculum source and symptoms would usually develop on the coleoptile (Shah and Bergstrom 1999). Our findings of P. nodorum on the roots could indicate that seedborne inoculum also is able to infect roots. Seedborne inoculum of Z. tritici is not considered as important, although in Argentina proof of natural seed infestation has been presented (Consolo et al. 2009). In the case of O. yallundae it has been shown that the ability of this fungus to colonize the plant depends on the pathogenicity of isolates. More pathogenic isolates of O. yallundae are able to penetrate the leaf sheaths causing stem infection whereas the less pathogenic are unable to spread beyond the leaf sheaths (Lucas et al. 2000).
F. poae, involved in the Fusarium Head Blight (FHB) complex, was detected in grains most frequently during this study. It is a relatively weak pathogen compared to F. culmorum and F. graminearum, which are also in the FHB complex, but it produces several mycotoxins, including trichothecenes, beauvericin and enniatins (Stenglein 2009). An increasing occurrence of F. poae has been observed in many countries in Europe (Xu et al. 2005). Dominance of F. poae among FHB pathogens increases the risk of grain contamination with a broad spectrum of mycotoxins produced by this fungus and indicates that this species can become more important in the future.
Microdochium bolleyi and Didymella exitalis, known as minor pathogens of wheat, were frequently detected. M. bolleyi, common on wheat and barley roots, can infect the plant causing discoloration on crown and roots, and some isolates can also reduce plant emergence and shoot dry weight (Gonzalez and Trevathan 2000). D. exitalis causes leaf scorch of cereals. There is no information about infection of grains by this fungus but our results demonstrate that Didymella exitalis was very common in the grains. The source of infection was probably air-borne ascospores produced by the fungus late in the season on senesced leaves (Cromey et al. 2004).
The grass pathogen Ophiosphaerella sp. causing spring dead spot disease in bermudagrass (Smiley et al. 2005) was found among the fungi colonizing wheat roots and grain. This is the first report of Ophiosphaerella sp. from wheat in Sweden. The role of Ophiosphaerella sp. in wheat is unknown but it has been shown that damage can occur in wheat after oats and in wheat monoculture (Kwaśna et al. 2010) suggesting that this fungus also can be a potential wheat pathogen.
Significant interactions between some pathogens were detected, particularly in the upper part of the stem. Most of the combinations of two pathogens showed positive relationships in which the presence of one pathogen could favour the occurrence of the other. Z. tritici and P. nodorum have similar life cycles and environmental requirements (Bockus et al. 2010) and it can explain the positive interaction observed between them in the stem samples. Negative interactions between these species and Fusarium spp. in the upper part of the stem could indicate that they compete in the colonization of the stem tissue. The opposite situation was observed in stem bases (Table 4). Z. tritici and P. nodorum are better adapted to infect leaf tissue than the stem base but it seems that in the presence of other pathogens these fungi are also able to colonize stem bases.
Positive interactions were also found in combinations of Fusarium spp. with F. poae and G. avenacea (anamorph F. avenaceum) which confirms that pathogenic Fusarium species often occur as species assemblages (Pettitt et al. 2003; Xu et al. 2005). The occurrence of and interactions between Fusarium species cannot be further resolved with the method used in our study.
In the stem bases a negative association was observed between M. nivale and O. yallundae, suggesting that these pathogens are antagonists or that conditions favouring one pathogen do not favour the other. The results are in contrast to our knowledge about these fungi. M. nivale, causing brown foot rot and snow mold, and O. yallundae, causing eyespot, have previously often been found together in the stem bases (Bateman et al. 2000; Matusinsky et al. 2008). In mixed infections it is not clear which of them is the primary or secondary colonizer (Matusinsky et al. 2008). In two fields from our study either M. nivale or O. yallundae dominated and they were found together only sporadically. In the third field M. nivale was the most frequent but mixed infections were observed more often compared to the other fields. These differences between fields influenced the log-linear analysis resulting in a negative interaction between M. nivale and O. yallundae when all fields were analyzed together.
Most of the wheat pathogens showed a significant positive interaction with saprophytic fungi meaning that the presence of non-pathogens favoured the colonization by pathogen, and/or vice versa. We found no indications that any of the saprophytic fungi could be regarded as potential antagonist preventing infections by pathogens. Antagonistic effect of saprophytic fungi inhabiting wheat against wheat pathogens has been observed frequently but only under experimental conditions (Liggitt et al. 1997; Müllenborn et al. 2008; Perelló et al. 2002). In the field the interactions between fungi colonizing the same tissue are more complex and influenced by many external factors, which sometimes can facilitate the growth of both pathogenic and non-pathogenic fungi.
We developed a reference database containing T-RFLP patterns of 67 fungal taxa, which were obtained after T-RFLP analysis of sequenced clones and isolates of fungi colonizing different parts of wheat. Although most of the fungal taxa collected in the reference library were found in the analyzed plant samples, unknown fungal profiles which did not match with patterns from the library were observed. These profiles can represent fungi rarely occurring on wheat and thereby unidentified by cloning or isolation and not included in the reference database.
The unknown fungal profiles can also represent isolates of fungal species already collected in the library but differing in sequences within the ITS region. The fungal ITS rDNA gene region contains both variable and highly conserved sequences (Thies 2007). Sequence variations in the ITS region within the same fungal species have been reported (Avis et al. 2006; Vujanovic et al. 2009). If the variation occurs in the restriction enzyme recognition site fragments of different length within the same species are obtained after cutting with restriction enzymes. This would result in different T-RFLP profiles for different individuals of the same species. The risk that some patterns will not be identified can be decreased by including as many T-RFLP profiles of one species as possible in the database (Dickie and FitzJohn 2007).
We could not identify fungi in 180 samples. The reason could be poor quality of restriction profiles obtained after T-RFLP. One of the criteria for the identification of fungal taxa in TRAMPR was the presence of peaks for minimum four out of six of the enzyme/primer combinations in a sample. Samples which did not fulfil this requirement could not be analysed by the program. Another reason for the unsuccessful identification of taxa could be that the obtained profile did not match with any of the fungal pattern from the library.
The internal transcribed spacer (ITS) region of the rDNA genes is commonly used for identification of fungal species and analysis of fungal communities (Allmer et al. 2006; Blixt et al. 2010; Dickie et al. 2002; Kwaśna et al. 2008; Lindahl et al. 2007). In the present study, however, the species Fusarium culmorum, F. graminearum and F. sporotrichioides as well as Cladosporium cladosporioides and C. tenuissimum could not be distinguished by analysis of the ITS region, with the restriction enzymes that were used, because of its low polymorphic nucleotide sequences. Very low variation in the sequence of the ITS region in the genus Fusarium has already been reported (Edel et al. 1997; Yli-Mattila et al. 2004). Recent phylogenetic studies of Fusarium have shown that the translation elongation factor 1α (TEF-1α) gene is a useful region for identification and distinguishing between species (Edel et al. 1997; Edel-Hermann et al. 2015; Kristensen et al. 2005). C. cladosporioides and C. tenuissimum are very closely related species (Bensch et al. 2010). Our results indicate high similarity between sequences of ITS regions resulting in difficulties in separation of those species by analysis of restriction profiles. Schubert et al. (2007) reported that within five genes (ITS, actin, calmodulin, translation elongation factor 1-α and histone H3) used for identification of species in the C. herbarum complex only ITS data did not provide any resolution.
The fungal population on organically grown winter wheat was very diverse. It consisted of fungi, which specifically colonized below- and above-ground parts of the plants. The composition of fungi changed during crop development but a pattern common for the whole crop could not be observed. However, for the particular plant part of wheat some patterns in the dynamics of fungal populations were found.
Among the fungi colonizing wheat some wheat pathogens were very common. They changed over time and also slightly between fields together with the other fungi found on the wheat. Most of the pathogens were specialized on plant part, a few of them were also found in unexpected places indicating the high adaptive ability of these fungi.
Significant interactions between pathogens and saprophytic fungi were observed but no evidence that any of the saprophytes could be considered as potential antagonists against pathogens was found.
References
Abdo, Z., Schuette, U. M. E., Bent, S. J., Williams, C. J., Forney, L. J., & Joyce, P. (2006). Statistical methods for characterizing diversity of microbial communities by analysis of terminal restriction fragment length polymorphisms of 16S rRNA genes. Environmental Microbiology, 8(5), 929–938.
Allmer, J., Vasiliauskas, R., Ihrmark, K., Stenlid, J., & Dahlberg, A. (2006). Wood-inhabiting fungal communities in woody debris of Norway spruce (Picea abies (L.) Karst.), as reflected by sporocarps, mycelial isolations and T-RFLP identification. FEMS Microbiology Ecology, 55(1), 57–67.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J. H., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25(17), 3389–3402.
Avis, P. G., Dickie, I. A., & Mueller, G. M. (2006). A ‘dirty’ business: testing the limitations of terminal restriction fragment length polymorphism (TRFLP) analysis of soil fungi. Molecular Ecology, 15(3), 873–882.
Bateman, G. L., Edwards, S. G., Marshall, J., Morgan, L. W., Nicholson, P., Nuttall, M., et al. (2000). Effects of cultivar and fungicides on stem-base pathogens, determined by quantitative PCR, and on diseases and yield of wheat. Annals of Applied Biology, 137(3), 213–221.
Bateman, G. L., & Kwaśna, H. (1999). Effects of number of winter wheat crops grown successively on fungal communities on wheat roots. Applied Soil Ecology, 13(3), 271–282.
Bensch, K., Groenewald, J. Z., Dijksterhuis, J., Starink-Willemse, M., Andersen, B., Summerell, B. A., et al. (2010). Species and ecological diversity within the Cladosporium cladosporioides complex (Davidiellaceae, Capnodiales). Studies in Mycology, 67, 1–94.
Blixt, E., Olson, A., Lindahl, B., Djurle, A., & Yuen, J. (2010). Spatiotemporal variation in the fungal community associated with wheat leaves showing symptoms similar to stagonospora nodorum blotch. European Journal of Plant Pathology, 126(3), 373–386.
Bockus, W. W., Bowden, R. L., Hunger, R. M., Morrill, W. L., Murray, T. D., & Smiley, R. W. (2010). Compendium of wheat diseases and pests.: APS Press, St. Paul
Colwell, R. K., & Coddington, J. A. (1994). Estimating terrestrial biodiversity through extrapolation. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 345(1311), 101–118.
Consolo, V. F., Albani, C. M., Beron, C. M., Salerno, G. L., & Cordo, C. A. (2009). A conventional PCR technique to detect Septoria tritici in wheat seeds. Australasian Plant Pathology, 38(3), 222–227.
Cromey, M. G., Butler, R. C., Mace, M. A., & Cole, A. L. J. (2004). Effects of the fungicides azoxystrobin and tebuconazole on Didymella exitialis, leaf senescence and grain yield in wheat. Crop Protection, 23(11), 1019–1030.
Culman, S. W., Bukowski, R., Gauch, H. G., Cadillo-Quiroz, H., & Buckley, D. H. (2009). T-REX: software for the processing and analysis of T-RFLP data. BMC Bioinformatics, 10, 171.
Culman, S. W., Gauch, H. G., Blackwood, C. B., & Thies, J. E. (2008). Analysis of T-RFLP data using analysis of variance and ordination methods: A comparative study. Journal of Microbiological Methods, 75(1), 55–63.
Dickie, I. A., & FitzJohn, R. G. (2007). Using terminal restriction fragment length polymorphism (T-RFLP) to identify mycorrhizal fungi: a methods review. Mycorrhiza, 17(4), 259–270.
Dickie, I. A., Xu, B., & Koide, R. T. (2002). Vertical niche differentiation of ectomycorrhizal hyphae in soil as shown by T-RFLP analysis. New Phytologist, 156(3), 527–535.
Edel-Hermann, V., Gautheron, N., Mounier, A., & Steinberg, C. (2015). Fusarium diversity in soil using a specific molecular approach and a cultural approach. Journal of Microbiological Methods, 111, 64–71.
Edel, V., Steinberg, C., Gautheron, N., & Alabouvette, C. (1997). Evaluation of restriction analysis of polymerase chain reaction (PCR)-amplified ribosomal DNA for the identification of Fusarium species. Mycological Research, 101, 179–187.
FitzJohn, R. G., & Dickie, I. A. (2007). TRAMPR: an R package for analysis and matching of terminal-restriction fragment length polymorphism (TRFLP) profiles. Molecular Ecology Notes, 7(4), 583–587.
Gardes, M., & Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes – application to identification of mycorrhizae and rusts. Molecular Ecology, 2(2), 113–118.
Gauch, H. G. (1992). Statistical analysis of regional yield trials: AMMI analysis of factorial designs. Amsterdam: Elsevier Science Publishers.
Gonzalez, M. S., & Trevathan, L. E. (2000). Identity and pathogenicity of fungi associated with root and crown rot of soft red winter wheat grown on the Upper Coastal Plain Land Resource Area of Mississippi. Journal of Phytopathology-Phytopathologische Zeitschrift, 148(2), 77–85.
Hovmøller, M. S., Sørensen, C. K., Walter, S., & Justesen, A. F. (2011). Diversity of Puccinia striiformis on cereals and grasses. Annual Review of Phytopathology, 49, 197–217.
Kristensen, R., Torp, M., Kosiak, B., & Holst-Jensen, A. (2005). Phylogeny and toxigenic potential is correlated in Fusarium species as revealed by partial translation elongation factor 1 alpha gene sequences. Mycological Research, 109, 173–186.
Kwaśna, H., Bateman, G. L., & Ward, E. (2008). Determining species diversity of microfungal communities in forest tree roots by pure-culture isolation and DNA sequencing. Applied Soil Ecology, 40(1), 44–56.
Kwaśna, H., Bateman, G. L., & Ward, E. (2010). Microbiota in wheat roots evaluated by cloning of ITS1/2 rDNA and sequencing. Journal of Phytopathology, 158(4), 278–287.
Larran, S., Perelló, A., Simón, M. R., & Moreno, V. (2002). Isolation and analysis of endophytic microorganisms in wheat (Triticum aestivum L.) leaves. World Journal of Microbiology and Biotechnology, 18(7), 683–686.
Larran, S., Perelló, A., Simón, M. R., & Moreno, V. (2007). The endophytic fungi from wheat (Triticum aestivum L.). World Journal of Microbiology and Biotechnology, 23(4), 565–572.
Lemańczyk, G., & Sadowski, C. K. (2002). Fungal communities and health status of roots of winter wheat cultivated after oats and oats mixed with other crops. BioControl, 47(3), 349–361.
Liggitt, J., Jenkinson, P., & Parry, D. W. (1997). The role of saprophytic microflora in the development of Fusarium ear blight of winter wheat caused by Fusarium culmorum. Crop Protection, 16(7), 679–685.
Lindahl, B. D., Ihrmark, K., Boberg, J., Trumbore, S. E., Högberg, P., Stenlid, J., et al. (2007). Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytologist, 173(3), 611–620.
Lindblad, M., Gidlund, A., Sulyok, M., Börjesson, T., Krska, R., Olsen, M., et al. (2013). Deoxynivalenol and other selected Fusarium toxins in Swedish wheat - Occurrence and correlation to specific Fusarium species. International Journal of Food Microbiology, 167, 284–291.
Lucas, J. A., Dyer, P. S., & Murray, T. D. (2000). Pathogenicity, host-specificity, and population biology of Tapesia spp., causal agents of eyespot disease of cereals. In J. A. Callow (Ed.), Advances in Botanical Research Incorporating Advances in Plant Pathology, Vol 33 (Vol. 33, pp. 225–258, Advances in Botanical Research).
Magurran, A. E. (1988). Ecological diversity and its measurement. Princeton, NJ, USA: Princeton University Press.
Matusinsky, P., Mikolasova, R., Spitzer, T., & Klem, K. (2008). Colonization of winter wheat stem bases by communities of pathogenic fungi. Cereal Research Communications, 36(1), 77–88.
Müllenborn, C., Steiner, U., Ludwig, M., & Oerke, E. C. (2008). Effect of fungicides on the complex of Fusarium species and saprophytic fungi colonizing wheat kernels. European Journal of Plant Pathology, 120(2), 157–166.
Nicolaisen, M., Justesen, A. F., Knorr, K., Wang, J., & Pinnschmidt, H. O. (2014). Fungal communities in wheat grain show significant co-existence patterns among species. Fungal Ecology, 11, 145–153.
Perelló, A., Simón, M. R., & Arambarri, A. M. (2002). Interactions between foliar pathogens and the saprophytic microflora of the wheat (Triticum aestivum L.) phylloplane. Journal of Phytopathology-Phytopathologische Zeitschrift, 150(4–5), 232–243.
Pettitt, T., Xu, X. M., & Parry, D. (2003). Association of Fusarium species in the wheat stem rot complex. European Journal of Plant Pathology, 109(7), 769–774.
Pfender, W. F., & Wootke, S. L. (1988). Microbial communities of Pyrenophora-infested wheat straw as examined by multivariate analysis. Microbial Ecology, 15(1), 95–113.
Roos, J., Hopkins, R., Kvarnheden, A., & Dixelius, C. (2011). The impact of global warming on plant diseases and insect vectors in Sweden. European Journal of Plant Pathology, 129(1), 9–19.
Shah, D. A., & Bergstrom, G. C. (1999). Epidemiology of seedborne Stagonospora nodorum: a case study of New York winter wheat. In M. van Ginkel, A. McNab, & J. Krupinsky (Eds.), The fifth international Septoria workshop, CIMMYT Mexico 1999 (pp 102–104)
Shivanna, M. B., Meera, M. S., & Hyakumachi, M. (1996). Role of root colonization ability of plant growth promoting fungi in the suppression of take-all and common root rot of wheat. Crop Protection, 15(6), 497–504.
Sieber, T., Riesen, T. K., Muller, E., & Fried, P. M. (1988). Endophytic fungi in four winter wheat cultivars (Triticum aestivum L.) differing in resistance against Stagonospora nodorum (Berk.) Cast. & Germ. = Septoria nodorum (Berk.) Berk. Journal of Phytopathology-Phytopathologische Zeitschrift, 122(4), 289–306.
Smiley, R. W., Dernoeden, P. H., & Clarke, B. B. (2005). Compendium of turfgrass diseases (3rd ed.): APS Press.
Stenglein, S. A. (2009). Fusarium poae: a pathogen that needs more attention. Journal of Plant Pathology, 91(1), 25–36.
Thies, J. E. (2007). Soil microbial community analysis using terminal restriction fragment length polymorphisms. Soil Science Society of America Journal, 71(2), 579–591.
Vujanovic, V., Mavragani, D., & Hamel, C. (2012). Fungal communities associated with durum wheat production system: A characterization by growth stage, plant organ and preceding crop. Crop Protection, 37, 26–34.
Vujanovic, V., Vidovic, S., Fernandez, M. R., Daida, P., & Korber, D. (2009). Whole-cell protein and ITS rDNA profiles as diagnostic tools to discriminate Fusarium avenaceum intraspecific variability and associated virulence. Canadian Journal of Microbiology, 55(2), 117–125.
Waern, P., & Sandström, M. (2007). Växtskyddsåret 2007. Dalarna, Gästrikland, Hälsingland, Uppland och Västmanlands län. Jordbruksinformation 15. Jönköping: Jordbruksverket.
White, T. J., Bruns, T. D., Lee, S., & Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In T. J. White, J. J. Sninsky, D. H. Gelfand, & M. A. Innin (Eds.), PCR Protocols a Guide to Methods and Applications (pp. 315–322). San Diego, USA: Academic Press.
Wiik, L. (2009). Yield and disease control in winter wheat in Southern Sweden during 1977-2005. Crop Protection, 28(1), 82–89.
Xu, X. M., Parry, D. W., Nicholson, P., Thomsett, M. A., Simpson, D., Edwards, S. G., et al. (2005). Predominance and association of pathogenic fungi causing Fusarium ear blight in wheat in four European countries. European Journal of Plant Pathology, 112(2), 143–154.
Yli-Mattila, T., Mach, R. L., Alekhina, I. A., Bulat, S. A., Koskinen, S., Kullnig-Gradinger, C. M., et al. (2004). Phylogenetic relationship of Fusarium langsethiae to Fusarium poae and Fusarium sporotrichioides as inferred by IGS, ITS, beta-tubulin sequences and UP-PCR hybridization analysis. International Journal of Food Microbiology, 95(3), 267–285.
Zadoks, J. C., Chang, T. T., & Konzak, C. F. (1974). A decimal code for the growth stages of cereals. Weed Research, 14, 415–421.
Acknowledgments
We wish to thank Ian A. Dickie from Landcare Research, Lincoln, New Zealand who adjusted the software TRAMPR to our data. We thank also Robert Bukowski from Cornell University, Ithaca, USA for help with T-REX software. We are grateful to Björn Lindahl for reading and commenting on the manuscript. The authors also thank M. Löfgren, M. Jonsson and D. Rönnlund for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Online Resource 1
T-RFLP pattern of Cladosporium and Fusarium species included in the reference database. Values are apparent fragment lengths (bp) obtained by using restriction enzymes Alu I, Cfo I, Taq I (PDF 27 kb)
Online Resource 2
The occurrence of the most common fungal taxa on winter wheat during one growing season analysed by logistic regression and shown as the odds of finding fungi in plant parts. The upper and lower 95 % confidence intervals are shown (PDF 48 kb)
Rights and permissions
About this article

Cite this article
Grudzinska-Sterno, M., Yuen, J., Stenlid, J. et al. Fungal communities in organically grown winter wheat affected by plant organ and development stage. Eur J Plant Pathol 146, 401–417 (2016). https://doi.org/10.1007/s10658-016-0927-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-016-0927-5