
Abstract
The restoration of mining wastelands, particularly in karst regions contaminated by heavy metals, is an environmental challenge in need of urgent attention. Soil microbes play a vital role in nutrient cycling and ecosystem recovery, yet the long-term evolution of soil microbial communities in such settings remains poorly understood. This study explored the dynamics and influencing factors of soil microbial communities during 35 years of natural restoration in abandoned manganese (Mn) mine areas in Guangxi Province, China. The results revealed that the concentrations of Mn, Cd, Zn, and Cu were significantly (p < 0.05) reduced by 80.4–85.3%, 55.3–70.0%, 21.0–38.1%, and 29.4–49.4%, respectively, in the mid-late restoration periods (R19 and R35) compared with R1. The α diversities of the bacterial and fungal communities significantly increased in the middle–late restoration periods (R19 and R35), indicating increased microbial diversity as restoration progressed. The bacterial community structure exhibited more pronounced changes than did the fungal community structure, with significant shifts observed in dominant phyla such as Proteobacteria, Actinobacteria, Acidobacteriota, and Ascomycota. Notably, the relative abundances of Rhizobiales, Burkholderiales, and Hypocreales increased gradually with succession. Co-occurrence network analysis revealed that bacterial interactions became stronger over time, whereas interactions between bacteria and fungi weakened. Mantel tests and partial least squares path modeling (PLS‒PM) identified soil pH, heavy metals (Mn, Cd, Zn, and Cu), and nutrients (SOM and TN) as key drivers shaping the microbial community composition. These factors were more strongly correlated with bacterial communities than with fungal communities, underscoring the different responses of microbial groups to environmental changes during natural restoration. These findings enhance our understanding of the ecological processes governing microbial community succession in heavy metal-contaminated soils undergoing natural restoration.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The process of metallic mineral mining inevitably destroys the topsoil layer and increases heavy metal (HM) pollution (Liu et al., 2018, 2020c). Many HMs (Mn, Cd, Pb, Zn, Cu, etc.) enter the topsoil and then may enter organisms through the food chain, posing a serious threat to human health and survival (Liu et al., 2020b; Nong et al., 2022). Therefore, there is an urgent need to remediate mining wastelands.
Natural restoration has been widely adopted as a highly effective strategy for rehabilitating degraded ecosystems (Hu et al., 2020; Yin et al., 2023). The success of natural restoration depends not only on the establishment of plant cover and soil physicochemical characteristics but also on the regeneration of soil microbial properties, which has received limited attention (Jiang et al., 2022; Zhang et al., 2016). Soil microorganisms play pivotal roles in numerous ecosystem processes, including nutrient cycling and energy transfer, and the development of stable soil systems (Singh et al., 2022; Zhao et al., 2021a). Previous investigations have demonstrated that natural restoration can increase the abundance and diversity of microbial communities through the augmentation of plant residue inputs and increases in soil organic C and N (Cai et al., 2022; Hu et al., 2020; Liu et al., 2020a; Zhang et al., 2021). Conversely, other studies have shown that natural restoration leads to a decrease in the biomass and/or richness of soil microbial communities (Zhao et al., 2021b). These inconsistent results may be attributed to various factors, including the heterogeneity of ecosystems, the type and duration of restoration efforts, and changes in the soil substrate (Hu et al., 2020, 2022). Moreover, studies on the natural restoration of abandoned manganese (Mn) mines are limited.
Soil microbial communities are primarily controlled by biotic and abiotic factors (Deng et al., 2020; Zhang et al., 2016), among which soil properties are considered vital (Cai et al., 2022). For example, a few studies have shown that total HMs such as Cd, Zn, Cu, Pb, and Mn can reduce microbial diversity and alter microbial compositions (Duan et al., 2021; Nong et al., 2022; Wu et al., 2022a; Yin et al., 2023). Changes in the quantity and quality of soil nutrients can greatly influence the composition of soil microbial communities. This phenomenon arises because various microbial groups employ distinct strategies for substrate utilization (Zhang et al., 2016). Soil pH has been widely recognized as a predominant factor driving soil microbial community dynamics (Deng et al., 2020; Hu et al., 2022). Therefore, identifying the biotic and abiotic factors regulating soil microbial community changes will contribute to a better understanding of the mechanisms driving soil microbial community succession.
The space-for-time substitution method is a commonly used and effective method for studying the succession patterns of ecosystems, including plant succession (Peng, 1994) and/or soil evolution (Cai et al., 2022; Hu et al., 2022; Zhang et al., 2016), over long time scales. Soil evolution provides insights into long-term changes in soil properties, nutrient cycling, and overall ecosystem functioning (Cai et al., 2022; Hu et al., 2022; Zhang et al., 2016). Several researchers have used the space-for-time substitution method to study the physicochemical properties and microbial recovery patterns of karst areas, forests and Loess Plateau soils (Cai et al., 2022; Hu et al., 2022; Jiang et al., 2021). Additionally, this method has been used in the restoration of rare earth element mines (Chao et al., 2016; Wei et al., 2019), metallic mines (Deng et al., 2020; Tong et al., 2017) and lignite tailing soil (Singh et al., 2022). Therefore, the space-for-time substitution method can be regarded as a practical approach for understanding and studying the regulators of ecological restoration. However, with respect to metallic mine restoration, previous studies have focused mainly on iron (Deng et al., 2020), copper (Tong et al., 2017), and rare earth elements (Chao et al., 2016; Wei et al., 2019) in mine wastelands, and related studies on Mn mine wasteland restoration have rarely been reported in the literature.
The Guangxi Zhuang Autonomous Region, a province in Southwest China with a population of 5012.68 million, lies in one of the world’s major karst regions and has the largest quantity and most extensive exploitation of Mn ore in China (Li et al., 2007; Liu et al., 2020c). The intensive mining of Mn leads to Mn toxicity in organisms, soil and water, which has adversely impacted local sustainable development and led to an urgent need for restoration (Li et al., 2019; Nong et al., 2022). The Daxin Mn mine is the largest and most representative Mn mining area in China. Previous studies have investigated Mn pollution in abandoned sites (Liu et al., 2024) and the migration and accumulation of Mn in dominant plants (Liu et al., 2020c). However, there has been no research on microbial succession patterns in Mn mine wastelands.
Therefore, in this study, we set up a space-for-time substitution experiment to investigate the long-term dynamic characteristics of soil Mn, Cd, Zn, and Cu concentrations; soil organic matter (SOM), total nitrogen (TN), total phosphorus (TP), available phosphorus (AP), potassium (K), calcium (Ca), sodium (Na), and magnesium (Mg); and microbial characteristics, including soil enzyme activities (invertase, alkaline phosphatase, protease, and urease) and, in particular, soil microbial community structure (bacteria, fungi) at different restoration ages. The stages considered were 1 and 3 years (representing the early restoration periods) and 19 and 35 years (representing the mid-late restoration periods), and the experiment was established at the Daxin Mn mine wasteland area in Guangxi Province, Southwest China. The main goals of this study were to (1) clarify how the compositions and diversities of the soil bacterial and fungal communities changed during the succession of Mn mines; (2) explore the changes in the characteristics of microbial species in successional sequences; and (3) identify the key factors that contribute to the changes in microbial community structure. This research provides scientific and theoretical references for Mn mine wasteland restoration.
Materials and methods
Study sites, experimental design, and sampling
The Daxin Mn Mine (22.908°–22.918° N; 106.683°–106.746° E) is located in the southern Guangxi Zhuang Autonomous Region, China, where it covers an area of 20.46 km2 and borders Vietnam. It is currently the largest surveyed Mn mine base in China and is known as the “Manganese Capital of China”.
The Daxin Mn Mine became operational in 1959 and has been in operation for more than 60 years. After complete exploitation, each pit is filled with waste rock and then overlaid with stockpiled topsoil, after which the area undergoes natural restoration. Therefore, the area presents sampling sites at different stages of restoration, providing an opportunity to study soil succession.
This region is well known for its spectacular karst landforms and is located within the subtropical monsoon climate zone, with an average annual temperature of 21.3 °C, an average annual sunshine duration of 1695 h, and an average annual rainfall of 1423.8 mm (Li et al., 2010). In summary, unique geological development and hot and rainy climatic conditions are favorable for the natural restoration of these Mn mining areas; these conditions also rapidly accelerate HM migration and diffusion, resulting in a pollution over a larger area.
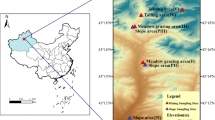
On the basis of preliminary field survey results, we used the space-for-time substitution method to select four sites in this study. At the time of the study, the selected areas had experienced natural restoration for 1, 3, 19, and 35 years, coded R1, R3, R19, and R35, respectively (Fig. 1; Table S1). In each sampling plot, debris, plant, and animal residues on the surface were removed before soil collection. To minimize variations within the field, 25–30 soil samples (0–20 cm depth) were taken at each sampling site using a soil auger (5 cm diameter) (Hu et al., 2022). The collected soils at each site were carefully mixed into a composite sample and then passed through a 2 mm sieve.

Appearances of the soil sampling sites. R1, R3, R19, and R35 represent soil sampling sites in abandoned areas of the Daxin Mn mine with restoration durations of 1, 3, 19, and 35 years, respectively
Each composite soil sample was divided into 3 parts and placed in a sterile bag. Ten grams of soil for microbial analysis was immediately placed in a − 20 °C dry ice box for preservation until subsequent DNA extraction. Five hundred grams of soil for soil enzyme analysis was stored in a 4 °C container, transported to the laboratory and stored in a refrigerator (4 °C). The remaining soil samples were air-dried for the determination of physicochemical properties.
Environmental parameter analysis
The soil pH was measured in a soil‒water mixture (1:2.5) using a pH meter (PHS-3C, China) (Yu et al., 2022). The soil sample (0.25 g) was digested with a 12 mL mixture of hydrochloric acid (HCl), nitric acid (HNO3), and perchloric acid (HClO4) at 3:1:1 (v/v) to obtain the digestion solution. The total concentrations of Mn, Zn, Cu, Cd, K, Ca, Na, and Mg were measured using an atomic absorption spectrometer (PinAAcle 900H, PerkinElmer, USA) (Agency, 2007; Liu et al., 2020b; Liu et al., 2020c; Sarojam & Chen 2011).
The total nitrogen (TN) and soil organic matter (SOM) contents were measured using an automated elemental analyzer (Vario MICRO cube, Elementar Instruments, Inc., Germany). The total phosphorus (TP) and available phosphorus (AP) contents were determined according to the methods described by Yu et al. (2022).
Soil enzyme activity measurement
The soil invertase, urease, protease, and alkaline phosphatase activities were determined using the 3,5-dinitrosalicylic acid method, indophenol colorimetric method, ninhydrin colorimetric method, and phenyl disodium phosphate colorimetric method, respectively (Liang et al., 2022; Małachowska-Jutsz & Matyja, 2019). All the chemical reagents used for soil determination were analytically pure.
DNA extraction and sequencing
Total soil DNA was extracted using the E.Z.N.A.® Soil Kit (Omega Bio-Tek, Norcross, GA, United States) following the manufacturer’s instructions. The DNA concentration and purity were measured with a NanoDrop2000 instrument (Thermo Fisher Scientific, USA), and the quality of the extracted DNA was evaluated using 1% agarose gel electrophoresis. The primers 515F (5’-GTGYCAGCMGCCGCGGTAA-3’) and 806R (5’-GGACTACNVGGGTWTCTAAT-3’) were used to amplify the 16S rRNA gene in the V4 region of the soil bacterial genome (Walters et al., 2016). The DNA in the ITS1‒ITS2 regions of the fungal genome was amplified by PCR using the primers ITS1F (5’-CTTGGTCATTTAGAGGAAGTAA-3’) and ITS2R (5’-GCTGCGTTCTTCATCGATGC-3’) (Adams et al., 2013). For each sample, three replicates were used. The PCR products were recovered using 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). The proteins were eluted with Tris–HCl and detected by 2% agarose electrophoresis. Detection and quantification were performed using QuantiFluor™-ST (Promega, USA). The paired-end (PE) library was constructed using the NEXTFLEX Rapid DNA-Seq Kit and by performing the following steps: (1) adapter ligation; (2) bead-based selection to remove self-ligated fragments; (3) enrichment of library templates using PCR amplification; and (4) bead recovery of PCR products to obtain the final library. Sequencing was performed using the Illumina MiSeq PE300 platform (Majorbio, Shanghai).
Sequence data processing
The original sequencing data were quality controlled using Fastp (version 0.19.6) to remove low-quality bases and adapter sequences. The initial paired-end raw read length was 250 bp. The total numbers of raw reads (2,933,826 and 2,933,826 for bacteria and fungi, respectively) were obtained. FLASH software (version 1.2.7) was used to merge the high-quality reads, resulting in the formation of longer contigs. Despite quality control and merging, PCR and sequencing errors might still have been present. To eliminate these errors and obtain accurate amplicon sequence variants (ASVs), which are considered more precise and comprehensive than operational taxonomic units (OTUs),
the data were further processed using DADA2. The species-level taxonomy of the ASVs was analyzed using the BLAST classifier in QIIME2, which is based on Sliva 16S rRNA (v 138) and the UNITE ITS reference database (version 8.0). To minimize the impact of sequencing depth on subsequent alpha diversity and beta diversity analyses, all the sample sequence numbers were standardized to the minimum number of sequences (100,415 and 123,031 for bacteria and fungi, respectively). The 16S rRNA and ITS gene sequences were submitted to the NCBI Sequence Read Archive (SRA) database with the numbers PRJNA1012899 and PRJNA1013054, respectively.
Statistical analysis
In our study, the data were processed using Microsoft Excel 2016 and SPSS (statistical package for the social sciences) 19.0. One-way analysis of variance (ANOVA) with Duncan’s multiple range test was used to determine the significance of the results (p < 0.05) (SPSS 19.0). Microbial alpha diversity analysis, principal coordinate analysis (PCoA) and microbial species variance analysis were conducted on the Majorbio Cloud platform. Based on the Bray‒Curtis distance, PCoA was used to assess overall differences in the soil microbial community structure. A Venn diagram was generated using the Venn package in R (version 4.2.2) to explore the distribution patterns of bacteria and fungi at the ASV level. The ggcor package in R (version 4.2.2) was used to depict the Mantel correlations between microbial communities and environmental factors. Both heatmaps and bar charts were drawn using Origin 2019b. Multiple linear regression analysis was used to determine the differences in the Shannon index, Chao1 index, NMDS1 score and soil properties. Pearson correlation coefficients were used to analyze the relationships between basic soil physical and chemical properties, soil enzyme activities and the relative abundances of soil microbes. Based on the reports of Ji et al. (2021) and Hu et al. (2022), genera with relative abundances greater than 1% were selected for further analysis, and the co-occurrence networks of the soil bacteria and fungi in the four restoration duration groups were created using Gephi version 0.9.7 software. A partial least squares path model (PLS‒PM) was constructed using SmartPLS-4 software to explore the direct and indirect effects of natural restoration, soil properties, diversity, and the bacterial and fungal communities on microbial function.
Results
Soil properties
As shown in Table 1, the highest HM concentration was found for Mn, followed by Zn, Cu, and Cd. All the HM concentrations exceeded the background levels in Guangxi Province, with particularly notable levels of Mn in R1 and R3; these levels were 192- and 162-fold higher than the provincial background values, respectively. The soil pH and HM concentrations decreased significantly (p < 0.05) over time. The pH in R35 was 8.06%, 11.93%, and 16.75% lower than that in R19, R3, and R1, respectively. The HM concentrations exhibited a pattern similar to that of the soil pH, i.e., R1 > R3 > R19 > R35. Compared with those in R1, the concentrations of Mn, Cd, Zn, and Cu in R3, R19 and R35 were reduced by 15.6%-85.3%, 19.2%-70.0%, 9.1%-38.1%, and 3.6%-49.4%, respectively. The soil SOM, TN, K, Na, and Mg contents were significantly (p < 0.05) higher in the middle to late restoration periods (R19 and R35) than in the early restoration periods (R1 and R3). In contrast, the soil Ca content showed the opposite trend, with the values in R3, R19, and R35 decreasing by approximately 14.18%, 57.07%, and 51.95%, respectively, compared with that in R1. In addition, the soil AP content increased and then decreased over time, with the highest value of 24.37 mg/kg being observed in R19, while the TP content exhibited no obvious trend over time.
Soil enzyme activities
The changes in soil enzyme activities are shown in Fig. 2. In general, the activities of the soil enzymes invertase, alkaline phosphatase, protease, and urease were significantly greater in the middle–late restoration periods (R19 and R35) than in the early restoration periods (R1 and R3). In addition, soil enzyme activities were significantly (p < 0.001, p < 0.01, or p < 0.05) negatively correlated with the HM contents, pH, and Ca and significantly (p < 0.001, p < 0.01, or p < 0.05) positively correlated with the SOM, TN, Na, and Mg contents (Fig. 7).

Variations in soil enzyme activities (invertase (A), alkaline phosphatase (B), protease (C), and urease (D)) across different durations of natural restoration in abandoned areas of the Daxin Mn mine in Guangxi Province, China. The values are the means ± SDs (n = 3). One-way ANOVA (analysis of variance) was performed for each parameter, and different lowercase letters above the bars within the same columns denote a significant difference among the restoration durations at p < 0.05
Soil microorganisms
Soil microbial community diversity
After quality filtering, all the rarefaction curves tended to be flat, indicating a reasonable sequencing depth (Fig. S1). A total of 1,415,557 valid bacterial gene sequences were obtained from 12 soil samples, and 23,981 ASVs were obtained by clustering. The taxonomic annotations revealed that the soil bacterial communities at sites with different restoration durations in the abandoned Daxin Mn mine areas included specimens from 54 phyla, 160 classes, 380 orders, 619 families, and 1172 genera.
In this study, α diversity indices such as the Chao1 and Shannon indices were used to assess microbial diversity over time (Fig. 3). In general, the soil bacterial community α diversity in the middle to late restoration periods (R19 and R35) was significantly (p < 0.05) greater than that in the early restoration period (R1 and R3). However, the difference in α diversity between R1 and R3 as well as that between R19 and R35 was not significant (p > 0.05) (Fig. 3A).

Variations in the alpha diversity of the soil microbial communities (A: bacteria, B: fungi) after different durations of natural restoration in abandoned areas of the Daxin Mn mine in Guangxi Province, China. The values are the means ± SDs (n = 3). One-way ANOVA (analysis of variance) was performed for each parameter, and different lowercase letters above the bars within the same columns denote a significant difference among the restoration durations at p < 0.05
A total of 1,654,388 valid fungal gene sequences were obtained from 12 soil samples, and 8766 ASVs were obtained by clustering. The taxonomic annotations revealed that the soil fungal communities at sites with different restoration durations in the abandoned Daxin Mn mine areas included 13 phyla, 51 classes, 130 orders, 326 families, and 819 genera. The Chao1 index of the soil fungal community increased (p < 0.05) and was significantly greater in the middle to late restoration period (R19 and R35) (p < 0.05) than in the early restoration period (R1 and R3). The Shannon index remained the same over time (Fig. 3B).
Similarly, with increasing restoration duration, the structures of the bacterial and fungal communities changed (p = 0.001) (Fig. 3). Axes 1 and 2 explained 46.24% and 20.93% of the variation in the bacterial β diversity at the ASV level, respectively, and R1, R3, R19, and R35 were distributed in different quadrants (Fig. 4A). The first two principal coordinates together explained 63.28% of the variation in the fungal β diversity at the ASV level. R1 was close to R3, and it was obviously separated from R19 and R35 along axis 2 (Fig. 4B), which fully explained the differences and similarities in microbial community diversity among the different groups of samples. The results indicated that the changes in the bacterial community structure were more obvious than those in the fungal community structure.

PCoA of the β diversity of soil bacteria (A) and fungi (B) after different durations of natural restoration in abandoned areas of the Daxin Mn mine in Guangxi Province, China. The percentages marked on the horizontal and vertical axes are the contributions of the principal components to explaining the differences in the ASVs
Soil microbial community composition
Analysis of microbial community composition and structure is a crucial step in exploring the potential dynamics of a community. In this study, Venn diagrams revealed differences in the ASVs of soil bacteria and fungi during natural restoration (Fig. S2). The number of shared bacterial ASVs was 323, accounting for 1.35% of the total bacterial ASVs. The unique ASVs at each site accounted for 60.37%, 63.10%, 66.98%, and 77.98% of the total bacterial ASVs. The number of shared fungal ASVs was 205, accounting for 2.34% of the total fungal ASVs, and the unique ASVs at each site accounted for 55.51%, 56.88%, 62.21%, and 70.27% of the total fungal ASVs.
As shown in Fig. 5, the phyla Proteobacteria (23.5–37.45%), Actinobacteria (17.58–25.44%), and Acidobacteriota (12.46–17.92%) dominated the soil bacterial communities, and the dominant fungal phyla at all four restoration stages were Ascomycota (38.61–66.55%) and Basidiomycota (9.57–44.8%) (Fig. 5, Table S2). Notably, the highest relative abundance of Actinobacteria was observed in R3, at 25.44%. The relative abundance of Acidobacteriota was greater in the middle–late restoration periods (R19 and R35) than in the early restoration periods (R1 and R3), although the increase was not significant. The relative abundance of Basidiomycota gradually decreased from R3-R35, and the relative abundance of Ascomycota significantly (p < 0.05) decreased over time. Additionally, the relative abundances of nondominant bacterial phyla, including Gemmatimonadota, Methylomirabilota, and Latescibacterota, increased in the middle–late restoration periods (R19 and R35) compared with those in the early restoration periods (R1 and R3). In contrast, Patescibacteria and WPS-2 decreased during the same periods.

Relative abundances of soil bacterial (A) and fungal (B) communities at the phylum level after different durations of natural restoration in abandoned areas of the Daxin Mn mine in Guangxi Province, China. Less than 1% of the community was categorized as “Others”
At the order level, for the bacterial community, the relative abundances of Rhizobiales (Proteobacteria), Burkholderiales (Proteobacteria) and Vicinamibacterales (Acidobacteriota) were greater in the later restoration periods than in the earlier restoration periods. The relative abundance of Acidobacteriales (Acidobacteriota) exhibited the opposite trend (Fig. S3A). For the fungal community, the relative abundances of Hypocreales (Ascomycota) and Agaricales (Basidiomycota) were greater in the middle–late restoration periods (R19 and R35) than in the early restoration periods (R1 and R3), whereas the relative abundance of Chaetothyriales (Ascomycota) decreased over time (Fig. S3B).
Microbial co-occurrence network analysis
To better explore the potential interactions between soil bacterial and fungal communities, a microbial co-occurrence network was constructed, and its topological characteristics were analyzed (Fig. 6, Table S4). In this study, the total number of edges, average degree, network density, and number of edges among bacteria in the soil microbial co-occurrence network were greater in the middle–late restoration periods (R19 and R35) than in the early restoration periods (R1 and R3), with increases of 13.32%, 17.18%, 21.74% and 7.59% in R35 compared with R1, respectively. Moreover, the modularity index and the number of edges showed opposite trends between bacteria and fungi. All the above results indicated that the relationships between bacteria became stronger over time, whereas the relationships between bacteria and fungi gradually weakened. Interestingly, the proportion of bacterial nodes was the highest in all the studied soil samples. Furthermore, the percentage of fungal nodes reached a maximum in R35, with a value of was 37.41%, indicating that natural restoration promoted the proliferation of fungi to some extent. Notably, the top 4 bacteria and 2 fungi in the network were assigned to Proteobacteria, Actinobacteria, Acidobacteria, Chloroflexi, Ascomycota, and Basidiomycota. Moreover, Rhizobiales, Burkholderiales, Hypocreales, and Pleosporales were identified as the top four orders in the co-occurrence network.

Co-occurrence networks and topological properties of the soil bacterial and fungal communities after different natural restoration periods in abandoned areas of the Daxin Mn mine in Guangxi Province, China (A–D). A connection represents a statistically significant (p < 0.01) Spearman correlation with a coefficient of at least 0.6. The size of each node is proportional to the number of connections. The colors of the nodes represent their classification at the phylum level
Relationships among soil properties, the microbial community and microbial functions
Multiple linear regression analysis was conducted to examine the correlations between soil properties, enzyme activities, and the diversity of microbial communities, which was represented by the Chao1 and Shannon indices and NMDS1 scores for bacteria and fungi (Table S3). The Chao1 index, Shannon index, and NMDS1 score for the bacteria as well as the Chao1 index and NMDS1 score for the fungi were significantly (p < 0.001, p < 0.01, or p < 0.05) correlated with the soil pH; Mn, Cd, Zn, Cu, SOM, TN, K, Ca, Na, and Mg contents; and alkaline phosphatase, protease, urease, and invertase activities. The TP content was significantly (p < 0.05) correlated with only the Chao1 index of the bacteria, and the C/N ratio was significantly (p < 0.05) correlated with only the Chao1 index and NMDS1 score of the fungi. In contrast, the Shannon index of the fungi was not significantly (p > 0.05) correlated with the soil physicochemical properties or enzyme activities.
To test whether differences in the microbial community structure might be explained by the environmental variables, a Mantel test was performed (Fig. 7, Table S5). The soil pH, Mn, Cd, Zn, Cu, SOM, TN, TP, Ca, K, Na, and Mg contents and C/N were significantly correlated with both the bacterial and fungal community compositions. Furthermore, the TN (bacteria, r = 0.53, p = 0.001 vs. fungi, r = 0.40, p = 0.002), SOM (bacteria, r = 0.69, p = 0.001 vs. fungi, r = 0.40, p = 0.007), and C/N (bacteria, r = 0.47, p = 0.004 vs. fungi, r = 0.33, p = 0.009) were more strongly correlated with the bacterial community composition than with the fungal community composition (Fig. 8).

Mantel test between microbial communities (order level) and soil environmental factors and enzyme activities. *p < 0.05, ** p < 0.01 and *** p < 0.001

Heatmap of the Pearson correlation coefficients between the relative abundances of bacteria and fungi at the phylum level (average relative abundance > 1%) (A) and order level (top 15) (B) and the physicochemical factors and enzyme activities. *: p < 0.05, **: p < 0.01, ***: p < 0.001
A PLS‒PM was used to infer the direct and indirect effects of long-term natural restoration of the Mn mine wasteland on the microbial community and its functionality (Fig. 9A, B). Restoration had significant positive effects on soil nutrients (0.942) and negative effects on pH (− 0.953) and HMs (− 0.781) (p < 0.001). Restoration, HMs (1.628, p < 0.001) and pH (− 1.944, p < 0.001) jointly and significantly altered the bacterial community. Restoration and soil nutrients jointly and significantly altered the bacterial diversity (1.526, p < 0.05) and community structure (0.613, p < 0.05). Restoration, HMs (1.632, p < 0.05) and soil nutrients (1.419, p < 0.01) jointly and significantly altered the fungal community. Moreover, the activities of enzymes related to the C, N, and P cycles were significantly affected by the bacterial and fungal communities. Specifically, C-cycling-related enzyme activity was affected by bacterial diversity (0.859, p < 0.05) and fungal community structure (0.752, p < 0.05). N cycling-related enzyme activity was affected by bacteria and fungi (bacterial diversity: − 0.492, p < 0.05; bacterial community structure: 1.397, p < 0.001; fungal community structure: 0.984, p < 0.05). P-cycling-related enzyme activity was significantly affected by bacterial (1.214, p < 0.001) and fungal (1.040, p < 0.01) community structure.

PLS‒PM analysis of the direct and indirect effects of natural restoration, soil properties, diversity, and bacterial (A) and fungal (B) communities on microbial function. The green and orange arrows indicate positive and negative causal flows, respectively (p < 0.05). The blue dashed arrows indicate that the coefficient is not significant (p > 0.05), and R2 indicates the variance in the dependent variable that is explained by the model. HM: Mn; Soil nutrient: SOM, TN, K, Na, and Mg; bacterial community and fungal community: NMDS axis 1 score; bacterial diversity and fungal diversity: Chao1 and Shannon indices; C-cycling: invertase; N-cycling: protease and urease; and P-cycling: alkaline phosphate. Significant path coefficients are indicated by *p < 0.05, ** p < 0.01 and *** p < 0.001
Discussion
It has been shown that natural restoration can activate the accumulation of HMs in plants and the transport of HMs from soil to plants, reduce HM concentrations, and increase soil nutrients and enzyme activities (Hu et al., 2022; Jiang et al., 2022). Our investigation confirmed that the concentration of HMs significantly decreased over time, which was consistent with the findings of other studies (Duan et al., 2021). One plausible explanation for this finding is that natural restoration enables the retention of certain plant species that are tolerant to and able to enrich HMs, leading to a reduction in HMs within the rhizosphere via plant absorption (Yu et al., 2022). In addition, the contents of soil nutrients (TN, SOM, K, Na, Mg, and C/N) and the activities of soil enzymes (invertase, alkaline phosphatase, protease, and urease) significantly increased (p < 0.05) in the middle–late restoration periods (R19 and R35) (Table 1, Fig. 2). These results were consistent with the findings of Cai et al. (2022), who reported that greatly increased soil nutrients in the surface soil layer during vegetation restoration were primarily attributable to a substantial input of litter residues and root exudates into the soil. Moreover, greater root biomass and soil organic matter also increase the activities of enzymes (Cui et al., 2018). Our findings support this idea, as soil enzyme activities were found to be significantly (p < 0.001, p < 0.01, or p < 0.05) positively correlated with SOM, TN, Na, and Mg (Fig. 7). The nonsignificant changes in the soil TP content may be because derived P is mainly released from the weathering of soil minerals, which is a long-term biological process (Hu et al., 2022).
Ecological soil functions are determined by soil microbial communities. Soil microbial diversity is an important aspect of soil ecosystem restoration in mining areas (Chang et al., 2022). Changes in aboveground vegetation types cause alterations in soil factors, leading to differences in bacterial and fungal diversities (Hu et al., 2022). In our study, bacterial diversity and the fungal Chao1 index significantly increased (p < 0.05) in the middle–late restoration periods (R19 and R35) (Fig. 3), which is similar to the findings of Liu et al. (2020a) and Hu et al. (2022). Previous studies have shown that the abundance of microorganisms increases with changes in forest succession, whereas diversity sometimes decreases during late succession, which may be due to differences in the adaptability of different microbial communities to changes in soil physical and chemical properties (Liu et al., 2020a). It is widely recognized that soil HMs, such as Mn, Cd, Zn, Cu, Pb, Cr, and As, are crucial factors influencing bacterial diversity and can decrease bacterial biomass and diversity and inhibit bacterial enzyme activities in HM-contaminated soils (Wu et al., 2022a; Yin et al., 2023). Our study supports the above perspective: Mn, Cd, Zn, and Cu were significantly negatively correlated with bacterial diversity (Table S3). In addition, soil nutrients are recognized as key predictors of bacterial diversity. Our findings revealed a significant positive correlation between changes in bacterial diversity, fungal richness, and soil nutrients (e.g., SOM, TN, K, Na, Mg, and Ca) (Table S5), which supports the conclusions of Dai et al. (2023). This positive correlation is explained by the finding that higher nutrients in the middle‒late restoration periods could provide more energy sources, promoting the growth and metabolic activities of microorganisms (Dai et al., 2023). Higher levels of plant diversity can lead to greater variations in the soil microenvironment, thereby fostering the growth of more rhizosphere and nonrhizosphere microbial communities (Duan et al., 2021). Additionally, rich soil nutrients typically result from high plant diversity and cover, which also promote the proliferation of soil microorganisms (Yin et al., 2023). Gagnon et al. (2020) reported that vegetation cover significantly enhances bacterial diversity in mining regions. Correspondingly, the significant increase in bacterial diversity and fungal richness during the middle‒late restoration periods (R19 and R35) could be partly attributed to improvements in plant diversity and coverage, which warrants further study in the future. However, there was no significant change in the fungal Shannon index, which may reflect a stronger influence on fungal richness than on fungal diversity. Therefore, in this study, the variations in the bacterial diversity and fungal richness in the Mn mine wasteland area over the different restoration durations can be collectively attributed to heavy metal pollution and soil nutrient availability.
PCoA revealed that the bacterial and fungal community structures changed significantly across the restoration stages (p = 0.001), which reflected the response of the microbial community structure to changes in natural vegetation. The change in bacterial community structure across the restoration durations was greater than that in fungal community structure (Fig. 4). Similar results were obtained by Liu et al. (2019), who reported that revegetation significantly impacted bacterial community development. Previous studies have suggested that the faster growth rate, better nutrient acquisition ability, and stronger adaptability of bacteria cause them to have a stronger response than fungi do in the early stages of natural restoration (Zhang et al., 2021). Moreover, bacteria can metabolize a wider range of compounds, which may explain their relative variability, whereas fungi are strongly dependent on the presence of host trees (Yin et al., 2023). We also found that the bacterial diversity changed significantly across the restoration stages. However, Hu et al. (2022) reported that the change in fungal community structure was more obvious than that in bacterial community structure, which may be related to differences in the mining areas, soil conditions, plant species and restoration durations. In conclusion, our results suggest that bacterial communities are more responsive than fungal communities to restoration in abandoned Daxin Mn mining areas.
We further analyzed microbial changes at different taxonomic levels. At the phylum level, Proteobacteria, Actinobacteria, and Acidobacteriota were dominant at all four natural restoration stages, which is consistent with the results of other studies on microbial communities in mining areas (Chang et al., 2022; Nong et al., 2022; Pereira et al., 2022), suggesting that these bacterial taxa play important roles in soil nutrient cycling and in reducing heavy metal toxicity. The significant increase in Actinobacteriota in R3 suggests that this species plays a pivotal role in the early restoration periods (Fig. 5A). Yan et al. (2020b) reported that Actinobacteria, which are gram-positive bacteria, have a high affinity for HMs due to their abundant heavy metal resistance genes. They can chelate and adsorb HMs through robust secondary metabolism, facilitating their transformation and translocation (Wu et al., 2022a). Additionally, Actinobacteria contribute to soil nutrient cycling by mineralizing organic matter, thereby promoting plant growth (Li et al., 2024). Moreover, we found that the relative abundance of Acidobacteriota was greater in the middle–late restoration periods (R19 and R35). Microbial nutritional strategies can help us understand our results. For example, Acidobacteriota are K-strategists (Jiang et al., 2021). Previous studies have indicated that among bacteria, an increase in easily decomposable organic matter, such as glucose and root exudates, in the soil leads to a rapid decline in K-strategists, which triggers an excitation effect that reduces soil organic matter decomposition (Morrissey et al., 2017). In contrast, the increase in K-strategist bacteria observed in our study suggests that organic matter decomposition slows as succession progresses, leading to a more stable ecosystem in the later restoration stages. Among the fungi, we found that Ascomycota and Basidiomycota were the dominant phyla, and the primary fungal phylum shifted from Ascomycota in CK to Basidiomycota in R3. The relative abundance of Basidiomycota gradually decreased from R3-R35 (Fig. 5B). Previous studies have demonstrated that Basidiomycota are essential for the decomposition of recalcitrant organic matter and that the content of recalcitrant organic matter in litter increases over time, resulting in an increase in the abundance of Basidiomycota (Jiang et al., 2021; Yan et al., 2020a). Moreover, Ascomycota and Basidiomycota were found to have negative and positive correlations with the soil C/N ratio, respectively. This suggests a shift in fungal communities from r- to K-strategists, and the increase in C/N resulting from plant species and litter may drive this transition in fungal communities (Yan et al., 2020a). Further research is needed to understand how plant community dynamics influence fungal community shifts and how these interactions impact soil restoration processes.
At a finer taxonomic scale, i.e., the order level, we observed notable increases in the relative abundances of Rhizobiales, Burkholderiales, and Vicinamibacterales in the later restoration periods. In contrast, the relative abundance of Acidobacteriales decreased (Fig. S3A). This result was consistent with the findings of Chang et al. (2022). Wu et al. (2022b) reported that Vicinamibacterales contains a gcd gene and pit transporter protein-encoding gene, which are closely related to soil P solubilization and plant P uptake processes; this finding could explain why Vicinamibacterales abundance was significantly and positively correlated with alkaline phosphatase activity (Fig. 8B). Vicinamibacterales abundance was not strongly correlated with AP (Fig. 8B), which may be due to the acidic nature of the soil in the study area, which is not conducive to the conversion of organic P to inorganic P (Wu et al., 2022b). Therefore, vegetation growth in the later periods of restoration may be limited by P. Furthermore, Zheng et al. (2023) reported that resistant microorganisms could adapt to HM-contaminated habitats and increase in abundance. Acidobacteriales have often been found to be abundant in soils contaminated by multiple HMs (Zheng et al., 2023), indicating that they are also HM tolerant or resistant. This could explain why its relative abundance in the early restoration stages was greater than that in the later restoration stages and was significantly positively correlated with HMs (Fig. 8B). Among the fungi, the relative abundances of Hypocreales and Agaricales increased in the middle‒late restoration periods (R19 and R35), whereas that of Chaetothyriales showed the opposite trend. Agaricales is an important order in the phylum Basidiomycota, and members of this order can decompose lignin and other materials through mycorrhizal associations and interactions with other stenotrophic fungi, gradually degrading organic matter into more stable SOM and providing a better soil environment for subsequent plant growth (Kushwaha et al., 2021). Undoubtedly, the changes in microbial abundance are not always consistent among soils in areas undergoing succession. For example, a study on the natural restoration of abandoned rare earth element mine sites demonstrated that the abundances of Alphaproteobacteria, Planctomycetes, Verrucomicrobia, Firmicutes, and Bacteroidetes significantly increased or decreased (Chao et al., 2016). The reasons for the differences in the microbial community structure are multifaceted and require further study, and those detected here may be related to differences in mining areas, soil properties, and plant species. In summary, natural restoration significantly changed the soil microbial community composition and increased the number of beneficial microorganisms in the Mn mine wasteland area.
Within certain ecological niches, microorganisms interact in various ways, such as through commensalism, competition, and predation (Hu et al., 2022; Singh et al., 2022); such interactions can be revealed through microbial co-occurrence analysis. In our study, a modularity index > 0.4 indicated that the network had modular characteristics (Xue et al., 2017). Rhizobiales, Burkholderiales, and Hypocreales were the top three orders, which is similar to the results of Chang et al. (2022), suggesting that these species perform important ecological functions. Many studies have reported that Rhizobiales and Burkholderiales can form rhizobia with legumes to fix nitrogen and can also form contaminant degradation repair systems with plant roots, thereby reducing the HM concentration (Ji et al., 2021; Wei et al., 2019). Kepler et al. (2017) showed that Hypocreales can produce a variety of beneficial compounds, such as antibiotics, insecticides and plant growth-promoting substances, that play positive roles in maintaining ecological balance. In addition, Žifčáková et al. (2011) reported that some species of Hypocreales also have the ability to produce extracellular enzymes that are involved in material cycling in the ecosystem. Thus, all three key taxa may play critical roles in Mn mine wasteland areas. In the future, the functions of relevant microorganisms will be further explored to enhance the understanding of ecological restoration mechanisms in mining areas. Our results also revealed changes in network properties during restoration. For example, the proportion of bacterial nodes in the co-occurrence network decreased as successional progressed, whereas the proportion of fungal nodes showed the opposite trend (Fig. 6, Table S4). This suggests that in the early stages of succession, bacteria dominate the pioneer groups, and as soil nutrients increase, competition for organic matter from fungi also increases. Research has suggested that secondary forest succession plays a role in fostering the growth of a robust root system and enhancing nutrient release, thereby increasing soil fungal activity and the complexity of the fungal community structure and increasing the resilience of the community to environmental change (Bai et al., 2020). In general, the more diverse and complex the microbial community in the soil is, the more stable the soil ecosystem becomes, leading to improved ecological functions and a more pronounced buffering capacity against external environmental changes. Notably, a lower ratio of bacteria/fungi in the soil contributes to ecosystem stability (Hu et al., 2022). This study revealed that during the natural recovery process, there were increases in the soil bacterial and fungal abundances coupled with a decrease in the bacteria/fungi ratio, suggesting that vegetation restoration could effectively increase the ecosystem stability of the Mn mine wasteland ecosystem. The change in the ratio of bacteria to fungi led to different contributions of microbial decomposition and metabolite synthesis to soil carbon and nitrogen metabolism, thus driving the dynamics of soil carbon and nitrogen (Wagg et al., 2019), which may have resulted in an imbalance of these two elements. Moreover, there were strong positive correlations and few negative correlations in the co-occurrence network, suggesting that microbes cooperate to fill similar ecological niches (Kost et al., 2023).
The microbial community may be influenced by a variety of changing environmental factors, including HM contamination and soil physicochemical properties (Yin et al., 2023). In this study, we found strong correlations between the soil pH and the compositions of the soil microbial communities (Fig. 7). A previous study indicated that pH had a significant effect on the microbial community and that the microbial abundance increased as the pH decreased (Xue et al., 2017). Jones et al. (2019) reported that a soil pH value between 5 and 5.8 can maximize microbial energy efficiency. Therefore, in this study, the microbial communities in the soil were altered by the decrease in pH, which improved the energy metabolism efficiency of the microorganisms. The PLS‒PM results revealed that pH significantly affected the bacterial community but had little effect on the fungal community, possibly because the pH range suitable for bacterial growth is narrow, whereas fungi have a wide pH range (Rousk et al., 2010). Soil pH changes also affect the forms of HMs present in soil (Yu et al., 2022). In our study, HMs significantly affected the microbial community composition, which is similar to the findings of Yin et al. (2023) and Nong et al. (2022). Yin et al. (2023) concluded that total HMs, such as Cd, Zn, Cu, Pb, and Mn, reduced the microbial biomass and diversity and drove changes in the microbial community composition in an abandoned polymetallic mine. High concentrations of HMs have harmful effects on microbial cell membranes and intracellular enzymatic activity (Yin et al., 2023). Moreover, the synergistic effects among HMs exacerbate the toxic impact on bacterial enzymatic activity (Yin et al., 2023). These effects may explain why HMs significantly and negatively affected the abundances of Gemmatimonadetes, Methylomirabilota, Latescibacterota, Rozellomycota, Mortierellomycota and Glomeromycota (Fig. 8A). Conversely, high contents of Mn increased the abundance of Ascomycota. Ascomycota has been reported to be highly tolerant to HMs and can mitigate the toxic effects of HMs through the precipitation of extracellular secretions and adsorption by the cell wall (Yu et al., 2022). Therefore, we hypothesize that HMs, especially Mn, were the most important factors affecting the microbial community composition in the Mn mining wasteland over time.
Many studies have shown that the composition of soil microbial communities in mining areas is highly sensitive to soil nutrients (Deng et al., 2020; Gagnon et al., 2020; Yin et al., 2023). Deng et al. (2020) reported that during vegetation restoration, the accumulation of soil nutrients is the most crucial factor influencing soil microbial communities. In this study, soil nutrients (e.g., SOM, TN, C/N, K, Na, Mg, and Ca) significantly affected the microbial communities (Fig. 7). This finding partially contrasts with previous observations, which identified TN, AN, and SOC as the primary factors driving changes in bacterial community structure (Jiang et al., 2021; Yin et al., 2023). The correlations between soil nutrients and soil microbial communities differ among soil systems. For example, Cai et al. (2022) reported that soil SOC, AP, and TP were strongly associated with soil microbial communities during vegetation restoration stages on the Loess Plateau of China. Chao et al. (2016) reported that soil nutrients, including TC, TN, TP, and AP, strongly influenced the bacterial community at an abandoned REE mine site. Additionally, the Mantel test revealed that soil nutrients (especially SOM and TN) were greater drivers of bacterial communities than fungal communities (Fig. 7), which aligns with the findings of Hu et al. (2021), suggesting that bacterial changes were more dependent on nutrient supply. Zheng et al. (2019) noted that individual and combined soil physicochemical properties have a stronger influence on bacterial and archaeal communities than on fungal communities. This is partly because bacterial communities can rapidly become more diverse by utilizing unstable carbon and nitrogen more efficiently than can fungal communities (Chen et al., 2020). Moreover, our results revealed a significant influence of bacterial and fungal communities on the activity of enzymes related to the C, N, and P cycles (Fig. 9). Many authors have demonstrated a positive correlation between microbial abundance and soil enzyme activities (Kumar et al., 2013). This phenomenon has been attributed to the production of these enzymes by living microbial cells (Alves de Castro Lopes et al., 2013). Bacteria play indispensable roles in the cycling of biogeochemical elements (Landa et al., 2016). The processing of soluble organic matter with varying reactivities in the carbon cycle is predominantly mediated by diverse bacterial types (Landa et al., 2016). An increase in fungal diversity can increase the decomposition of plant litter and complex organic matter (Van Der Heijden et al., 1998), thereby potentially enhancing soil nutrient cycling. Microorganisms also actively participate in soil processes by secreting various compounds, thereby regulating the C, N, and P cycles in the soil. The aforementioned elucidation of the relationships among soil physicochemical properties, the microbial community, and microbial function enhances our comprehension of soil processes.
Conclusion
Our findings indicated that natural restoration effectively reduced HM pollution and enhanced soil fertility at abandoned Mn mine sites. With succession, bacterial diversity and community changes showed greater sensitivity, which was more closely related to the soil nutrient requirements. These findings indicate that bacteria played a more pronounced role in driving nutrient cycling during Mn mine wasteland restoration. In the future, Rhizobiales, Burkholderiales, Vicinamibacterales, Agaricales, and Hypocreales could be directly inoculated into the soil in Mn mining areas to investigate their impacts on plant growth and physiology. This study provides a new starting point for elucidating microbial response mechanisms in the ecological restoration of mining areas in karst regions, enhancing our understanding of the ability of microbial communities to colonize soil ecosystems through natural bioremediation. Furthermore, the response mechanisms of indigenous microorganisms responsible for biodegradation in the soils of Mn mining areas, including their stress resistance and the molecular mechanisms involved in their response processes, require further investigation.
Data availability
The 16S rRNA and ITS gene sequences were submitted to the NCBI Sequence Read Archive (SRA) database with the numbers PRJNA1012899 and PRJNA1013054, respectively.
References
Adams, R. I., Miletto, M., Taylor, J. W., & Bruns, T. D. (2013). Dispersal in microbes: Fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. The ISME Journal, 7, 1262–1273.
Agency, U.S.E.P., 2007. Method 7000B: Flame atomic absorption spectrophotometry.
Bai, Y., Zha, X., & Chen, S. (2020). Effects of the vegetation restoration years on soil microbial community composition and biomass in degraded lands in Changting County, China. Journal of Forestry Research, 31, 1295–1308.
Cai, X., Zhang, D., Wang, Y., Diao, L., Cheng, X., Luo, Y., An, S., & Yang, W. (2022). Shift in soil microbial communities along ~160 years of natural vegetation restoration on the Loess Plateau of China. Applied Soil Ecology, 173, 104394.
Chang, Y., Chen, F., Zhu, Y., You, Y., Cheng, Y., & Ma, J. (2022). Influence of revegetation on soil microbial community and its assembly process in the open-pit mining area of the Loess Plateau. China. Front Microbiol, 13, 992816.
Chao, Y., Liu, W., Chen, Y., Chen, W., Zhao, L., Ding, Q., Wang, S., Tang, Y. T., Zhang, T., & Qiu, R. L. (2016). Structure, variation, and co-occurrence of soil microbial communities in abandoned sites of a rare earth elements mine. Environmental Science and Technology, 50, 11481–11490.
Chen, J., Wang, P., Wang, C., Wang, X., Miao, L., Liu, S., Yuan, Q., & Sun, S. (2020). Fungal community demonstrates stronger dispersal limitation and less network connectivity than bacterial community in sediments along a large river. Environmental Microbiology, 22, 832–849.
Cui, Y., Fang, L., Guo, X., Wang, X., Zhang, Y., Li, P., & Zhang, X. (2018). Ecoenzymatic stoichiometry and microbial nutrient limitation in rhizosphere soil in the arid area of the northern Loess Plateau, China. Soil Biology and Biochemistry, 116, 11–21.
Dai, Z., Guo, X., Lin, J., Wang, X., He, D., Zeng, R., Meng, J., Luo, J., Delgado-Baquerizo, M., & Moreno-Jiménez, E. (2023). Metallic micronutrients are associated with the structure and function of the soil microbiome. Nature Communications, 14, 8456.
de Castro, A., Lopes, A., Gomes de Sousa, D. M., Chaer, G. M., dos Reis, B., Junior, F., Goedert, W. J., & de Carvalho Mendes, I. (2013). Interpretation of microbial soil indicators as a function of crop yield and organic carbon. Soil Science Society of America Journal, 77, 461–472.
Deng, J., Bai, X., Zhou, Y., Zhu, W., & Yin, Y. (2020). Variations of soil microbial communities accompanied by different vegetation restoration in an open-cut iron mining area. Science of the Total Environment, 704, 135243.
Duan, R., Lin, Y., Zhang, J., Huang, M., Du, Y., Yang, L., Bai, J., Xiang, G., Wang, Z., & Zhang, Y. (2021). Changes in diversity and composition of rhizosphere bacterial community during natural restoration stages in antimony mine. PeerJ, 9, e12302.
Gagnon, V., Rodrigue-Morin, M., Tremblay, J., Wasserscheid, J., Champagne, J., Bellenger, J.-P., Greer, C. W., & Roy, S. (2020). Vegetation drives the structure of active microbial communities on an acidogenic mine tailings deposit. PeerJ, 8, e10109.
Hu, L., Li, Q., Yan, J., Liu, C., & Zhong, J. (2022). Vegetation restoration facilitates belowground microbial network complexity and recalcitrant soil organic carbon storage in southwest China karst region. Science of the Total Environment, 820, 153137.
Hu, P., Xiao, J., Zhang, W., Xiao, L., Yang, R., Xiao, D., Zhao, J., & Wang, K. (2020). Response of soil microbial communities to natural and managed vegetation restoration in a subtropical karst region. Catena, 195, 104849.
Hu, W., Huang, L., He, Y., Liu, Y., Liu, Y., Kong, Z., Wu, L., & Ge, G. (2021). Soil bacterial and fungal communities and associated nutrient cycling in relation to rice cultivation history after reclamation of natural wetland. Land Degradation & Development, 32, 1287–1300.
Ji, L., Si, H., He, J., Fan, L., & Li, L. (2021). The shifts of maize soil microbial community and networks are related to soil properties under different organic fertilizers. Rhizosphere, 19, 100388.
Jiang, S., Xing, Y., Liu, G., Hu, C., Wang, X., Yan, G., & Wang, Q. (2021). Changes in soil bacterial and fungal community composition and functional groups during the succession of boreal forests. Soil Biology and Biochemistry, 161, 108393.
Jiang, X., Guo, Y., Li, H., Li, X., & Liu, J. (2022). Ecological evolution during the three-year restoration using rhizosphere soil cover method at a Lead-Zinc tailing pond in Karst areas. Science of the Total Environment, 853, 158291.
Jones, D. L., Cooledge, E. C., Hoyle, F. C., Griffiths, R. I., & Murphy, D. V. (2019). pH and exchangeable aluminum are major regulators of microbial energy flow and carbon use efficiency in soil microbial communities. Soil Biology and Biochemistry, 138, 107584.
Kepler, R. M., Maul, J. E., & Rehner, S. A. (2017). Managing the plant microbiome for biocontrol fungi: Examples from Hypocreales. Current Opinion in Microbiology, 37, 48–53.
Kost, C., Patil, K. R., Friedman, J., Garcia, S. L., & Ralser, M. (2023). Metabolic exchanges are ubiquitous in natural microbial communities. Nature Microbiology, 8, 2244–2252.
Kumar, S., Chaudhuri, S., & Maiti, S. (2013). Soil dehydrogenase enzyme activity in natural and mine soil-a review. Middle-East Journal of Scientific Research, 13, 898–906.
Kushwaha, P., Neilson Julia, W., Barberán, A., Chen, Y., Fontana Catherine, G., Butterfield Bradley, J., & Maier Raina, M. (2021). Arid ecosystem vegetation canopy-gap dichotomy: Influence on soil microbial composition and nutrient cycling functional potential. Applied and Environmental Microbiology, 87, e02780-e2720.
Landa, M., Blain, S., Christaki, U., Monchy, S., & Obernosterer, I. (2016). Shifts in bacterial community composition associated with increased carbon cycling in a mosaic of phytoplankton blooms. The ISME Journal, 10, 39–50.
Li, J., Jia, Y., Dong, R., Huang, R., Liu, P., Li, X., Wang, Z., Liu, G., & Chen, Z. (2019). Advances in the mechanisms of plant tolerance to manganese toxicity. Int J Mol Sci, 20, 5096.
Li, J., Yin, R., Luo, Y., Lu, Y., & Zhang, L. (2010). Assessment of Heavy Metal Contamination of Soils in Daxin Manganese Mine Guangxi. Environmental Science & Technology, 33, 183–185.
Li, M. S., Luo, Y. P., & Su, Z. Y. (2007). Heavy metal concentrations in soils and plant accumulation in a restored manganese mineland in Guangxi, South China. Environmental Pollution, 147, 168–175.
Li, Z., Fang, F., Wu, L., Gao, F., Li, M., Li, B., Wu, K., Hu, X., Wang, S., Wei, Z., Chen, Q., Zhang, M., & Liu, Z. (2024). The microbial community, nutrient supply and crop yields differ along a potassium fertilizer gradient under wheat–maize double-cropping systems. Journal of Integrative Agriculture. https://doi.org/10.1016/j.jia.2024.01.031
Liang, X., Li, Y., Tang, S., Shi, X., Zhou, N., Liu, K., Ma, J., Yu, F., & Li, Y. (2022). Mechanism underlying how a chitosan-based phosphorus adsorbent alleviates cadmium-induced oxidative stress in Bidens pilosa L and its impact on soil microbial communities: A field study. Chemosphere, 295, 133943.
Liu, G.-Y., Chen, L.-L., Shi, X.-R., Yuan, Z.-Y., Yuan, L. Y., Lock, T. R., & Kallenbach, R. L. (2019). Changes in rhizosphere bacterial and fungal community composition with vegetation restoration in planted forests. Land Degradation & Development, 30, 1147–1157.
Liu, J., Jia, X., Yan, W., Zhong, Y., & Shangguan, Z. (2020a). Changes in soil microbial community structure during long-term secondary succession. Land Degradation & Development, 31, 1151–1166.
Liu, K., Fan, L., Li, Y., Zhou, Z., Chen, C., Chen, B., & Yu, F. (2018). Concentrations and health risks of heavy metals in soils and crops around the Pingle manganese (Mn) mine area in Guangxi Province, China. Environmental Science and Pollution Research, 25, 30180–30190.
Liu, K., Huang, X., Wei, Y., & He, P. (2024). Heavy metal pollution assessment and soil property evolution under a natural ecological restoration process in a manganese wasteland in China. Restoration Ecology, 32, e14017.
Liu, K., Li, C., Tang, S., Shang, G., Yu, F., & Li, Y. (2020b). Heavy metal concentration, potential ecological risk assessment and enzyme activity in soils affected by a lead-zinc tailing spill in Guangxi. China. Chemosphere, 251, 126415.
Liu, K., Zhang, H., Liu, Y., Li, Y., & Yu, F. (2020c). Investigation of plant species and their heavy metal accumulation in manganese mine tailings in Pingle Mn mine, China. Environmental Science and Pollution Research International, 27, 19933–19945.
Małachowska-Jutsz, A., & Matyja, K. (2019). Discussion on methods of soil dehydrogenase determination. International Journal of Environmental Science and Technology, 16, 7777–7790.
Morrissey, E. M., Mau, R. L., Schwartz, E., McHugh, T. A., Dijkstra, P., Koch, B. J., Marks, J. C., & Hungate, B. A. (2017). Bacterial carbon use plasticity, phylogenetic diversity and the priming of soil organic matter. The ISME Journal, 11, 1890–1899.
Nong, H., Liu, J., Chen, J., Zhao, Y., Wu, L., Tang, Y., Liu, W., Yang, G., & Xu, Z. (2022). Woody plants have the advantages in the phytoremediation process of manganese ore with the help of microorganisms. Science of the Total Environment, 863, 160995.
Peng, S. (1994). Studies on Succession of plant Community II. methods for dynamics research(summary). Ecologic Science, 2, 117–119.
Pereira, APd. A., Mendes, L. W., Oliveira, F. A. S., Antunes, J. E. L., Melo, V. M. M., & Araujo, A. S. F. (2022). Land degradation affects the microbial communities in the Brazilian Caatinga biome. Catena, 211, 105961.
Rousk, J., Bååth, E., Brookes, P. C., Lauber, C. L., Lozupone, C., Caporaso, J. G., Knight, R., & Fierer, N. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. The ISME Journal, 4, 1340–1351.
Sarojam, P., & Chen, J. (2011). Atomic Absorption, SPICE, p. 19.
Singh, P., Jain, K. R., Lakhmapurkar, J., Gavali, D., Desai, C., & Madamwar, D. (2022). Microbial community structure and functions during chronosequence-based phytoremediation programme of Lignite tailing soil. Environmental Technology & Innovation, 27, 102447.
Tong, J., Miaowen, C., Juhui, J., Jinxian, L., & Baofeng, C. (2017). Endophytic fungi and soil microbial community characteristics over different years of phytoremediation in a copper tailings dam of Shanxi, China. Science of the Total Environment, 574, 881–888.
Van Der Heijden, M. G., Klironomos, J. N., Ursic, M., Moutoglis, P., Streitwolf-Engel, R., Boller, T., Wiemken, A., & Sanders, I. R. (1998). Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature, 396, 69–72.
Wagg, C., Schlaeppi, K., Banerjee, S., Kuramae, E. E., & van der Heijden, M. G. (2019). Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nature Communications, 10, 1–10.
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., Humphrey, G., Parada, A., Gilbert, J. A., Jansson, J. K., Caporaso, J. G., & Fuhrman, J. A. (2016). Improved bacterial 16S rRNA gene (V4 and V4–5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. Msystems, 1, e00009-00015.
Wei, Z., Hao, Z., Li, X., Guan, Z., Cai, Y., & Liao, X. (2019). The effects of phytoremediation on soil bacterial communities in an abandoned mine site of rare earth elements. Science of the Total Environment, 670, 950–960.
Wu, B., Luo, H., Wang, X., Liu, H., Peng, H., Sheng, M., Xu, F., & Xu, H. (2022a). Effects of environmental factors on soil bacterial community structure and diversity in different contaminated districts of Southwest China mine tailings. Science of the Total Environment, 802, 149899.
Wu, X., Rensing, C., Han, D., Xiao, K.-Q., Dai, Y., Tang, Z., Liesack, W., Peng, J., Cui, Z., & Zhang, F. (2022b). Genome-resolved metagenomics reveals distinct phosphorus acquisition strategies between soil microbiomes. Msystems, 7, e01107-01121.
Xue, L., Ren, H., Li, S., Leng, X., & Yao, X. (2017). Soil bacterial community structure and co-occurrence pattern during vegetation restoration in karst rocky desertification area. Frontiers in Microbiology, 8, 2377.
Yan, B., Sun, L., Li, J., Liang, C., Wei, F., Xue, S., & Wang, G. (2020). Change in composition and potential functional genes of soil bacterial and fungal communities with secondary succession in Quercus liaotungensis forests of the Loess Plateau, western China. Geoderma, 364, 114199.
Yan, C., Wang, F., Liu, H., Liu, H., Pu, S., Lin, F., Geng, H., Ma, S., Zhang, Y., & Tian, Z. (2020b). Deciphering the toxic effects of metals in gold mining area: Microbial community tolerance mechanism and change of antibiotic resistance genes. Environmental Research, 189, 109869.
Yin, Y., Wang, X., Hu, Y., Li, F., & Cheng, H. (2023). Soil bacterial community structure in the habitats with different levels of heavy metal pollution at an abandoned polymetallic mine. Journal of Hazardous Materials, 442, 130063.
Yu, F., Tang, S., Shi, X., Liang, X., Liu, K., Huang, Y., & Li, Y. (2022). Phytoextraction of metal(loid)s from contaminated soils by six plant species: A field study. Science of the Total Environment, 804, 150282.
Zhang, C., Liu, G., Xue, S., & Wang, G. (2016). Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Soil Biology and Biochemistry, 97, 40–49.
Zhang, M., Dong, L., Wang, Y., Bai, X., Ma, Z., Yu, X., & Zhao, Z. (2021). The response of soil microbial communities to soil erodibility depends on the plant and soil properties in semiarid regions. Land Degradation & Development, 32, 3180–3193.
Zhao, J., Ma, J., Yang, Y., Yu, H., Zhang, S., & Chen, F. (2021a). Response of soil microbial community to vegetation reconstruction modes in mining areas of the loess plateau. China. Front Microbiol, 12, 714967.
Zhao, J., Ma, J., Yang, Y., Yu, H., Zhang, S., & Chen, F. (2021b). Response of soil microbial community to vegetation reconstruction modes in mining areas of the Loess Plateau. China. Frontiers in Microbiology, 12, 714967.
Zheng, Q., Hu, Y., Zhang, S., Noll, L., Böckle, T., Dietrich, M., Herbold, C. W., Eichorst, S. A., Woebken, D., & Richter, A. (2019). Soil multifunctionality is affected by the soil environment and by microbial community composition and diversity. Soil Biology and Biochemistry, 136, 107521.
Zheng, X., Zhong, Z., Xu, Y., Lin, X., Cao, Z., & Yan, Q. (2023). Response of heavy-metal and antibiotic resistance genes and their related microbe in rice paddy irrigated with treated municipal wastewaters. Science of the Total Environment, 896, 165249.
Žifčáková, L., Dobiášová, P., Kolářová, Z., Koukol, O., & Baldrian, P. (2011). Enzyme activities of fungi associated with Picea abies needles. Fungal Ecology, 4, 427–436.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (42267005) and the Guangxi Key Natural Science Foundation (Guangdong-Guangxi Joint Fund Project) (2022GXNSFDA080008). We thank Yongming Wei and Pingxian He for assisting with this study.
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by XH, YH and QL. The first draft of the manuscript was written by XH. ZL and KL edited the manuscript. KL provided fund support. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
I would like to declare on behalf of my coauthors that the work described is original research that has not been published previously and is not under consideration for publication elsewhere, in whole or in part. All the authors listed have approved the enclosed manuscript. We declare that we have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, X., Hong, Y., Li, Q. et al. Characteristics and driving forces of the soil microbial community during 35 years of natural restoration in abandoned areas of the Daxin manganese mine, China. Environ Geochem Health 46, 413 (2024). https://doi.org/10.1007/s10653-024-02204-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10653-024-02204-y