
Summary
To evaluate the potential gastric pH-dependent drug-drug interaction (DDI), safety and tolerability of famitinib co-administered with omeprazole in healthy subjects. Twenty healthy subjects were enrolled in a single-center, single-arm, open-label, fixed-sequence study. Famitinib was administered as a single oral 25 mg under a fasting condition on day 1, omeprazole (40 mg once daily) was given on days 10–14, concomitantly with famitinib on day 15, and for the follow-up 7 additional days (days 16–22). Blood samples were collected for the pharmacokinetic analysis of famitinib and its metabolite SHR116637 following each famitinib dose. Safety and tolerability were assessed during the whole progress via clinical laboratory tests. The least-squares geometric mean ratios (GMRs) (90% CI) of Cmax, AUC0-t and AUC0-∞ for famitinib combined with omeprazole to famitinib alone were 0.989 (0.953, 1.027), 0.956 (0.907, 1.007) and 0.953(0.905, 1.005) respectively. For the metabolite SHR116637, their GMRs (90% CI) of the above parameters were 0.851 (0.786, 0.920), 0.890 (0.838, 0.946)and 0.887 (0.835, 0.943), indicating the absence of significant differences in the parameters. During the treatment period, 9(45%) subjects reported 16 treatment emergent adverse events (TEAE), among which 6 subjects (30%) reported 9 TEAEs and 1 subject (5%) reported 1 TEAE during famitinib or omeprazole administered alone respectively, 5 subjects (25.0%) reported 6 TEAEs during in the combined administration phase. Omeprazole did not have a significant influence on the pharmacokinetics (PK) of famitinib and SHR116637, and the safety profile was good upon co-administration. ClinicalTrials.gov identifier NCT 05,041,920.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The oral administration route of tyrosine kinase inhibitors (TKIs) offers effectiveness in both solid and hematologic malignancies and is convenient for the patients, however, despite these advantages, it also causes a highly relevant new problem. Of recently approved orally administered cancer therapeutics, > 50% are characterized as having pH-dependent solubility [1,2,3]. The poor and variable pH-dependent solubility, together with other variable pharmacokinetic factors, contribute to a significant patient variability in plasma levels and exposure. Next to other factors, a majority of cancer patients frequently take acid–reducing agent (ARA) to alleviate gastroesophageal symptoms, thereby raising the potential for a gastric pH-dependent drug interaction [1]. This type of DDI may have detrimental effects on the efficacy of TKIs, with major clinical impacts described for some orally administrated targeted therapies (erlotinib, gefitinib, pazopanib, palbociclib), and conflicting results with many others, including nilotinib or dasatinib [1, 4, 5]. Long-term suppression of gastric acidity could decrease the absorption of certain major oral anticancer drugs with pH-dependent solubility and result in subsequent failure of therapy. To address this, guidelines are provided by the FDA and the European Medicines Agency (EMA) that recommend studying the DDI between pH-dependent drugs and ARAs.
Famitinib (famitinib-malate, SHR1020, Fig. 1A) is a novel and potent multi-targeted receptor TKI that targets at c-kit, vascular endothelial growth factor receptor 2 and 3 (VEGFR-2 and 3), platelet-derived growth factor receptor (PD-GFR), FMS-like tyrosine kinase-3 receptor (FLT3) and protooncogene tyrosine kinase receptor (RET), as shown in Fig. 1 [6, 7]. Due to its anti-angiogenic effect, it was effective against metastatic renal cell carcinoma, non-small cell lung cancer and metastatic breast cancer [8,9,10]. Clinical trials of famitinib in combination with the concurrent medication or chemoradiotherapy also showed its good antitumor abilities against other solid tumors such as metastatic urothelial carcinoma, advanced immunomodulatory triple-negative breast cancer, advanced nasopharyngeal carcinoma and gastric cancer, etc. [6, 11,12,13,14,15,16,17]. A phase I study showed that famitinib had favorable PK characteristics and was generally well-tolerated. The major circulating metabolite SHR116637 was the formation of N-desethyl famitinib (Fig. 1B), which is pharmacodynamically active but exhibits a lower potency than the parent drug [18]. Within the dosing range of 4–27 mg, the increase in Cmax and AUC0–24 h for famitinib and SHR116637 were proportional to the increase in dose level. The plasma level of SHR116637 is approximately equivalent to 3.6% of that of the parent drug, and both famitinib and SHR116637 were slowly removed from circulation [6]. After administration for 28 days, the degrees of famitinib accumulation in vivo were significantly lower than sunitinib and the major side effects were noted in terms of neutrocytopenia, thrombocytopenia and diarrhea, with particularly less severe fatigue and thrombocytopenia [6]. These toxicities had no significant accumulation while treatment proceeded, however, the common adverse events (AEs) of gastrointestinal reactions, such as nausea and diarrhea, needed ARAs and gastric mucosal protective to alleviate these AEs.

Chemical structures of Famitinib A and the metabolite SHR116637 B
According to the FDA guidance’s decision tree on the evaluation of gastric pH-dependent drug interactions, DDI studies with ARAs are required if the drug dissolution is too low to determine the effect of pH on drug solubility or the solubility of the drug at pH 6.0–6.8 is less than dose divided by 250 mL [2]. Famitinib has been classified as a BCS class IV drug (low solubility, low permeability) by the FDA. The results of in vitro solubility test showed that the solubility of famitinib was 85, 140 and 8 μg/mL in the medium system of pH 1.0, 4.5, 6.8 respectively. The magnitude of solubility change with increasing pH occurs at a pH of 6.0–6.8. It fits the above criteria, so it is necessary to explore the effects of pH on the PK of famitinib. Among all therapeutic agents, PPIs are the most prevalent and most potent ARAs and with daily use produce a marked and sustained duration of acid suppression [1, 2]. A prospective study in four French Comprehensive Cancer Centers, more than a quarter of cancer patients used PPIs, mostly on a daily basis and in the long term [19]. As PPIs generally have a longer duration of suppression effect on gastric acid secretion than H2 blockers and antacids do, they are expected to interfere with the intestinal absorption of TKIs to a greater extent [4]. In this paper, omeprazole, was therefore chosen for the study of famitinib with an PPI. We aim to update the potential gastric pH-dependent drug interactions between omeprazole and famitinib in healthy subjects, as well as to ascertain the safety of co-administration of famitinib and omeprazole.
Method
Study design
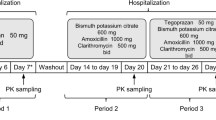
The screening was performed from day-7 to day-1. Eligible subjects were admitted to the Phase I clinical trial ward on day-1, provided a light diet in the evening, and then fasted for 10 h. On day 1, each subject was administered famitinib as a single oral 25 mg dose. Blood samples were collected before administration (within 1 h), 1, 2, 3, 4, 5, 6, 7, 8, 12, 24, 48, 72, 96, 144 and 192 h after dosing. On day 10 through 22, subjects were orally daily administered 40 mg of omeprazole at least 0.5 h before breakfast, with the exception of day 15. On day 15, famitinib (25 mg) was administered concomitantly with omeprazole (40 mg), and the collection of the blood samples was the same as that on day 1. During the study period, all drugs were administered with approximately 240 mL water under a fasting condition, on day 1 and day 15, water was forbidden within 1 h before and after the study drug administration, and food was to be avoided within 4 h after administration. On day 23, all subjects were discharged after examination in the morning. Subjects returned to the research center for follow-up or telephone follow-up from day 28 to day 30. A safety assessment was performed during the entire test period. A flowchart of this study is presented in Fig. 2.

The study design of the evaluation of gastric pH-dependent drug interactions between famitinib and omeprazole
Participants
Informed consent was signed before the trial, healthy male and female subjects aged between 18 and 45 (inclusive), of which no less than 1/3 are female subjects. The bodyweight of male subjects was ≥ 50 kg, and that of females was ≥ 45 kg; The body mass index should range between 19–28 kg/m2. Creatinine clearance (CLCr) ≥ 80 mL/min. Key exclusion criteria included QTcF > 470 ms for women or > 450 ms for men, Any history of dysphagia or any gastrointestinal disease that affects drug absorption, uncontrolled peptic ulcer, colitis, pancreatitis, etc. Full eligibility criteria are included in the Supplementary Methods.
Formulations
Jiangsu Hengrui Pharmaceuticals produced and supplied famitinib capsules (specification: 25 mg/capsule, Lot: 200906NS). Omeprazole magnesium enteric-coated tablets (specification: 20 mg/tablet, Lot: SAMU) were also provided by Jiangsu Hengrui Pharmaceuticals.
PK assessment
The plasma concentration of famitinib and SHR116637 was determined by liquid chromatography-tandem mass spectrometry (LC–MS/MS). The analytical method was developed and validated to meet the standard operating procedure established by the sponsor. The concentration range of calibration standards for famitinib and SHR116637 was both 0.05–100 ng/mL. In each analytical batch, the number of quality control samples (QC) accounts for more than 5% of the total number of samples, and at least two samples at each concentration level per time. For famitinib, the inter-run precision was 2.9–4.1%, while the inter-run accuracy ranged between 1.3–3.1%. For the metabolite SHR116637, the inter-run precision and accuracy were 1.6–3.4% and -2.7–1.1%, respectively.
Safety assessment
Safety was monitored by measurements of vital signs (blood pressure, heart rate and temperature), physical examination clinical laboratory tests and 12-lead electrocardiogram. Tolerability was assessed by recording adverse events (AEs). Details of any AEs were recorded, including the AEs types, incidence, severity (graded according to NCI-CTCAE5.0), onset and end time, serious AEs, correlation with the test drug, and outcomes.
Pharmacokinetic and Statistical Analysis
The PK parameters of above analytes were calculated using a standard noncompartmental analysis method (NCA) by Phoenix WinNonlin (Pharsight Corporation 8.3 or higher). The main evaluation indices were Cmax (maximum plasma concentration), the area under the curve of plasma concentration–time from zero to the last measurable concentration calculated by the linear trapezoid method (AUC0 −t), and the area under the curve of blood concentration from zero to infinity (AUC0−∞). The secondary evaluation indices were Tmax, elimination halflife (t1/2), apparent clearance rate (CL/F), and apparent volume of distribution (Vz/F). In addition, CL/F and Vz/F were not applicable to the metabolite SHR116637.
Statistical analysis was carried out using SAS v 9.4 (SAS Institute, Cary, NC, USA). Descriptive statistics and lists of PK parameters of the analyte were conducted and mean concentration–time curves were plotted. After natural log transformation, a mixed-effect model was used to fit PK parameters. Based on this model, the drugs were considered as fixed effects and the volunteers as random effects. The GMRs (co-administration of famitinib and omeprazole, and administration of famitinib alone) and their 90% CIs were estimated.
Results
Subject demographics
Among the 20 Chinese subjects, there were 13 males and 7 females; 18 were of Han nationality and 2 were of other nationalities. The median age was 24.5 years (range 19–37 y), and the average height (± SD) was 168.38 ± 8.291 cm. The average body weight (± SD) was 69.09 ± 10.337 kg, and the average body mass index (± SD) was 24.26 ± 2.333 kg/m2.
PK analysis
The main PK parameters of famitinib and its metabolite SHR116637 between famitinib alone and co-administration with omeprazole were showed in Tabel 1. No clinically significant difference was observed in the Cmax, AUC0-t and AUC0-∞ of famitinib between alone monotherapy and co-administration with omeprazole. The GMRs of Cmax, AUC0-t and AUC0-∞ of famitinib were 98.9%, 95.6% and 95.3%, respectively. The exposure of the metabolite SHR116637 was slightly decreased when famitinib was co-administered with omeprazole, with the Cmax, AUC0-t and AUC0-∞ of SHR116637 decreased by 14.9%, 11.0% and 11.3% upon co-administration. The GMRs of Cmax, AUC0-t and AUC0-∞ for SHR116637 were 85.1%, 89.0% and 88.7%, respectively. Evaluation of drug interactions between famitinib and omeprazole was shown in Tables 1 and 2. The mean plasma concentration–time curve is shown as Fig. 3. The plasma concentrations of famitinib were similar over time, and there was no significant change in PK parameters between administration of famitinib alone and co-administration with omeprazole.

The mean plasma concentration versus time profile for famitinib A and SHR116637 B after oral administration of famitinib 25 mg with and without omeprazole
Safety analysis
All the 20 subjects enrolled in this study completed 2 times of famitinib and 13 times of omeprazole administration as planned. A total of 9 (45.0%) subjects had 16 AEs, all of which were treatment-emergent adverse events (TEAEs) at grade 1 severity. No adverse events of grade 2 or above were reported. Among the total 16 AEs, 9 TEAEs occurred in 6 subjects (30.0%) in the single administration phase of famitinib; 1 subject (5.0%) had 1 TEAE in the single administration of omeprazole; At the stage of famitinib combined with omeprazole, 5 subjects (25.0%) had 6 TEAEs. A summary of the above TEAEs was shown in Table 3. 5 (25.0%) subjects had 10 TEAEs related to famitinib including alanine aminotransferase increased (10%), blood triglycerides increased (10%), gamma-glutamyl transferase increased (5%), basophil count increased (5%), white blood cells urine positive (5%), glucose urine present (5%), and blood glucose increased (5%). 2 (10.0%) subjects had 2 TEAEs related to omeprazole including gamma-glutamyl transferase increased (5%) and increased heart rate. All adverse events occurred during the study period were recovered / resolved at the end of the study.
Disscusion
Early assessment of pH-dependent DDIs for TKIs of poorly soluble and weakly acidic compounds offers various advantages for patient safety. Retrospective data suggest that TKI plasma concentration is decreased in patients receiving concomitant PPIs or H2 antagonists therapy with subsequently poorer oncologic outcomes, such as crizotinib, dasatinib, erlotinib, gefitinib, lapatinib and pazopanib, and recommended avoiding concomitant use of [20]. However, another recent systematic review and meta-analysis of the use of gastric-acid suppressants and oral anticancer treatments supported the evidence of a possible negative impact of such combinations [21]. To our knowledge, this is the first study to assess the PK and safety effects of omeprazole on the potent TKIs famitinib as well as its major metabolite SHR116637 in healthy subjects. Our findings suggested that omeprazole did not significantly impact the PK properties of both famitinib and SHR116637, demonstrating good safety on co-administration.
Key factors in the design of a pH-dependent DDI study include study population, selection of ARAs, type of crossover design (randomized or single-sequence), and the dose/dosing regimen etc. [2]. Previous clinical trials have demonstrated that famitinib showed linear dose-related pharmacokinetic characteristics in the dosing range of 4–27 mg. The recommended dose for phase II clinical trials is 25 mg. Hence, for the safety evaluation, the dose of famitinib was selected to be 25 mg. The main PK parameters were similar in terms of Cmax, AUC and t1/2 between the patients with advanced solid cancer and healthy subjects. Food intake was unlikely to impact on the PK of famitinib [6]. As a result, the study conducted under the fasted condition as it was likely to represent the worst-case scenario and can help guide cancer patients' treatment. For the selection of ARAs, omeprazole was chosen for 1) PPIs generally have a longer duration of suppression effect on gastric acid secretion than H2 blockers and antacids do, and are expected to interfere with the intestinal absorption of WBDs to a greater extent and a worst-case scenario in the in vivo evaluation of the pH effect. 2) Famitinib was mainly metabolized by CYP3A4/5 and CYP1A1/2, Furthermore, famitinib is a weak inhibitor of CYP3A4, but it’s unlikely to affect CYP3A4 due to a single dose at 25 mg (0.22 μM) in this study. As one of the most commonly used PPIs in clinic, omeprazole gives its high affinity for CYP2C19 and moderate affinity for CYP3A4, However, no obvious effect was detected in the present study, and it was widely used in the gastric pH-dependent drug interaction of TKIs such as erlotinib and sorafenib which were also metabolized by CYP3A4 [4, 22]. In addition, a self-control study was also used to overcome the influence of enzyme differences between individuals. The common dose of omeprazole is 20 mg qd, which can achieve maximum suppression of gastric acid within ~ 4 days, and the expected effect of a 40-mg dose follows a similar time course [20]. Therefore, a second dose of famitinib was administered after 5 days omeprazole administration to ensure that subjects achieved maximum inhibition of gastric-acid secretion.
Compared with famitinib single-dose administration, the geometric mean of AUC0-∞ was slightly reduced when famitinib was co-administered with omeprazole (1417.927 vs. 1351.939 h·ng/mL, decreased by approximately 4.7%), along with the Cmax, Tmax, t1/2, CL/F and Vz/F did not significantly differ between the two phases. The least squares GMRs of Cmax, AUC0-t and AUC0-∞ (90% CIs) of famitinib combined with omeprazole to famitinib alone were 0.989 (0.953, 1.027), 0.956 (0.907, 1.007) and 0.953 (0.905, 1.005) respectively, indicating the absence of significant differences in AUC0-t, AUC0-∞ and Cmax of famitinib when compared with famitinib alone. The metabolite SHR116637 data was consistent with the reduced absorption famitinib, and the metabolic ratio remained similarly small for both treatment arms with 0.075 versus 0.081 for famitinib and famitinib plus omeprazole, respectively. The median Tmax of famitinib metabolite SHR116637 was 5.00 h and 6.50 h, indicating that the peak time of famitinib metabolite SHR116637 was slightly prolonged after omeprazole combined with famitinib. The Cmax, AUC0-t and AUC0-∞ decreased by 14.9%, 11.0% and 11.3% for famitinib and famitinib plus omeprazole, respectively. The least squares GMRs of Cmax, AUC0-t and AUC0-∞ (90% CIs) of SHR116637 between coadministration group and alone group were 0.851 (0.786, 0.920), 0.890 (0.838, 0.946)and 0.887 (0.835, 0.943) respectively. Except the lower limit for the SHR116637 GMR of Cmax (90% CIs) is 78.6%, the least squares GMRs of AUC0-t, AUC0-∞ and Cmax (90% CIs) of both famitinib and SHR116637 are all in the range of 80%-125%. Compared the “no concomitant PPIs” versus “concomitant PPIs” based on their clinical characteristics, the exposure of metabolite SHR116637 is approximately equivalent to 9.47% and 8.72% of that of the parent drug, so it has little effect on the PK of famitinib.
According to one completed phase-I clinical trial, the most common AEs of famitinib included neutrocytopenia, thrombocytopenia and diarrhea. In some cases, it can also result in elevation of blood lipids and glucose [6, 18]. In our study, we also assessed the safety profile of combination therapy in the present study, compared with the above AEs, most AEs observed in the present study was mild, such as increased gamma-glutamyl transferase, increased basophil count, increased heart rate, alanine aminotransferase, blood glucose increased, blood triglycerides elevated, the positive urine test for leukocyte-esterase and sugar. During the treatment period, a total of 6 (30.0%) subjects had 9 AEs during famitinib alone, and 1 (5.0%) subject had 1 AE during the mutiple dose of omeprazole, while 5 (25.0%) subjects had 6 AEs during famitinib combined with omeprazole. Less severe and less frequent side effects were noted after the co-administration of omeprazole and famitinib compared with the single phase of famitinib, revealing the safety and tolerability of famitinib and omeprazole coadministration in clinical settings. Co-administration of famitinib and omeprazole was associated with good safety.
Conclusions
To conclude, the effect of PPIs on the efficacy of certain anticancer agents, particularly TKIs, is a major issue in daily practice. In this opinion paper, although the famitinib has pH-dependent solubility in vitro, the PPI omeprazole had minimal effect on the PK of famitinib and SHR116637 in healthy subjects. Therefore, famitinib as a formulated tablet can be administered with or without PPIs, such as omeprazole. In addition, interactions caused by other factors involved in absorption, apart from the pH effect, need to be considered during drug development on a case-by-case basis.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Raoul JL, Edeline J, Simmet V, Moreau-Bachelard C, Gilabert M, Frenel JS (2022) Long-Term Use of Proton Pump Inhibitors in Cancer Patients: An Opinion Paper. Cancers (Basel) 14(5).https://doi.org/10.3390/cancers14051156
Zhang L, Wu F, Lee SC, Zhao H, Zhang L (2014) pH-dependent drug-drug interactions for weak base drugs: potential implications for new drug development. Clin Pharmacol Ther 96(2):266–277.https://doi.org/10.1038/clpt.2014.87
Herbrink M, Nuijen B, Schellens JH, Beijnen JH (2015) Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat Rev 41(5):412–422.https://doi.org/10.1016/j.ctrv.2015.03.005
Kletzl H, Giraudon M, Ducray PS, Abt M, Hamilton M, Lum BL (2015) Effect of gastric pH on erlotinib pharmacokinetics in healthy individuals: omeprazole and ranitidine. Anticancer Drugs 26(5):565–572.https://doi.org/10.1097/CAD.0000000000000212
Pape E, Michel D, Scala-Bertola J, Schiestel T, Harle A, Bouchet S, Contet A, Pochon C, Gambier N (2016) Effect of esomeprazole on the oral absorption of dasatinib in a patient with Philadelphia-positive acute lymphoblastic leukemia. Br J Clin Pharmacol 81(6):1195–1196.https://doi.org/10.1111/bcp.12895
Zhou A, Zhang W, Chang C, Chen X, Zhong D, Qin Q, Lou D, Jiang H, Wang J (2013) Phase I study of the safety, pharmacokinetics and antitumor activity of famitinib. Cancer Chemother Pharmacol 72(5):1043–1053. https://doi.org/10.1007/s00280-013-2282-y
Mu X, Ma J, Zhang Z, Zhou H, Xu S, Qin Y, Huang J, Yang K, Wu G (2015) Famitinib enhances nasopharyngeal cancer cell radiosensitivity by attenuating radiation-induced phosphorylation of platelet-derived growth factor receptor and c-kit and inhibiting microvessel formation. Int J Radiat Biol 91(9):771–776.https://doi.org/10.3109/09553002.2015.1062574
Zhang W, Zhou AP, Qin Q, Chang CX, Jiang HY, Ma JH, Wang JW (2013) Famitinib in metastatic renal cell carcinoma: a single center study. Chin Med J (Engl) 126(22):4277–4281
Qu YY, Zhang HL, Guo H, Luo H, Zou Q, Xing N, Xia S, Sun Z, Zhang X, He C et al (2021) Camrelizumab plus Famitinib in Patients with Advanced or Metastatic Renal Cell Carcinoma: Data from an Open-label, Multicenter Phase II Basket Study. Clin Cancer Res 27(21):5838–5846.https://doi.org/10.1158/1078-0432.CCR-21-1698
Zhang M, Quan H, Fu L, Li Y, Fu H, Lou L (2021) Third-generation EGFR inhibitor HS-10296 in combination with famitinib, a multi-targeted tyrosine kinase inhibitor, exerts synergistic antitumor effects through enhanced inhibition of downstream signaling in EGFR-mutant non-small cell lung cancer cells. Thorac Cancer 12(8):1210–1218.https://doi.org/10.1111/1759-7714.13902
Qu YY, Sun Z, Han W, Zou Q, Xing N, Luo H, Zhang X, He C, Bian XJ, Cai J et al (2022) Camrelizumab plus famitinib for advanced or metastatic urothelial carcinoma after platinum-based therapy: data from a multicohort phase 2 study. J Immunother Cancer 10(5). https://doi.org/10.1136/jitc-2021-004427
Chen Q, Tang L, Liu N, Han F, Guo L, Guo S, Wang J, Liu H, Ye Y, Zhang L et al (2018) Famitinib in combination with concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma: a phase 1, open-label, dose-escalation Study. Cancer Commun (Lond) 38(1):66. https://doi.org/10.1186/s40880-018-0330-z
Chen L, Jiang YZ, Wu SY, Wu J, Di GH, Liu GY, Yu KD, Fan L, Li JJ, Hou YF et al (2022) Famitinib with camrelizumab and nab-paclitaxel for advanced immunomodulatory triple-negative breast cancer (FUTURE-C-PLUS): an open-label, single-arm, phase 2 trial. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-21-4313
Xia L, Peng J, Lou G, Pan M, Zhou Q, Hu W, Shi H, Wang L, Gao Y, Zhu J et al (2022) Antitumor activity and safety of camrelizumab plus famitinib in patients with platinum-resistant recurrent ovarian cancer: results from an open-label, multicenter phase 2 basket study. J Immunother Cancer 10(1). https://doi.org/10.1136/jitc-2021-003831
Thomas T (2021) Camrelizumab plus famitinib to treat RCC. Nat Rev Urol 18(10):576. https://doi.org/10.1038/s41585-021-00517-6
Xu RH, Shen L, Wang KM, Wu G, Shi CM, Ding KF, Lin LZ, Wang JW, Xiong JP, Wu CP et al (2017) Famitinib versus placebo in the treatment of refractory metastatic colorectal cancer: a multicenter, randomized, double-blinded, placebo-controlled, phase II clinical trial. Chin J Cancer 36(1):97. https://doi.org/10.1186/s40880-017-0263-y
Ge S, Zhang Q, He Q, Zou J, Liu X, Li N, Tian T, Zhu Y, Gao J, Shen L (2016) Famitinib exerted powerful antitumor activity in human gastric cancer cells and xenografts. Oncol Lett 12(3):1763–1768. https://doi.org/10.3892/ol.2016.4909
Xie C, Zhou J, Guo Z, Diao X, Gao Z, Zhong D, Jiang H, Zhang L, Chen X (2013) Metabolism and bioactivation of famitinib, a novel inhibitor of receptor tyrosine kinase, in cancer patients. Br J Pharmacol 168(7):1687–1706. https://doi.org/10.1111/bph.12047
Raoul JL, Guerin-Charbonnel C, Edeline J, Simmet V, Gilabert M, Frenel JS (2021) Prevalence of Proton Pump Inhibitor Use Among Patients With Cancer. JAMA Netw Open 4(6):e2113739. https://doi.org/10.1001/jamanetworkopen.2021.13739
van Leeuwen RW, van Gelder T, Mathijssen RH, Jansman FG (2014) Drug-drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol 15(8):e315–326. https://doi.org/10.1016/S1470-2045(13)70579-5
Indini A, Petrelli F, Tomasello G, Rijavec E, Facciorusso A, Grossi F, Ghidini M (2020) Impact of Use of Gastric-Acid Suppressants and Oral Anti-Cancer Agents on Survival Outcomes: A Systematic Review and Meta-Analysis. Cancers (Basel) 12(4). https://doi.org/10.3390/cancers12040998
Svedberg A, Vikingsson S, Vikstrom A, Hornstra N, Kentson M, Branden E, Koyi H, Bergman B, Green H (2019) Erlotinib treatment induces cytochrome P450 3A activity in non-small cell lung cancer patients. Br J Clin Pharmacol 85(8):1704–1709. https://doi.org/10.1111/bcp.13953
Acknowledgements
This work was supported by Jiangsu Hengrui Co. Ltd. The authors thank all the patients who participated in this study and their families, as well as all the investigators and site staff who made the study possible.
Funding
This project was also supported by the National Natural Science Foundation of China (No. 81503340), and the National Natural Science Foundation of Jiangsu Province (No. BK20150645), and the Project of scientific research plan for drug supervision, Grant/Award Number: 202106. We thank staff involved.
Author information
Authors and Affiliations
Contributions
Linlin Hu and Huiping Wang contributed to the study conception and design. Material preparation, data collection and analysis were performed by Linlin Hu, Mingmin Cai, Wei Qian, Lu Tang, Qiuyue Sun and Ting Dou. The first draft of the manuscript was written by Linlin Hu and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Research involving human participants and/or animals
This study was performed in line with the principles of the Declaration of Helsinki. The study protocol was approved (approval number, 2021ZDSYLL195-P01) by the Ethics Committee of the Zhongda Hospital, Medical School, Southeast University (Nanjing, China).
Conflict of interest statement
The authors declare no conflicts of interest. The author reports no conflicts of interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, L., Cai, M., Qian, W. et al. Phase I study to evaluate of the gastric pH-dependent drug interaction between famitinib and the proton pump inhibitor omeprazole in healthy subjects. Invest New Drugs 40, 1274–1281 (2022). https://doi.org/10.1007/s10637-022-01299-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-022-01299-3