
Summary
Background The Japanese Pharmaceuticals and Medical Devices Agency (PMDA) was established in 2004. Since then, various pieces of legislation, notices, and guidelines have been issued, and the regulatory approval pathways for domestic drugs have been diversified. However, the effects of these measures have not been fully examined. We examined the impact of these measures on the approval of antineoplastic drugs and the design of pivotal clinical trials for efficacy assessment by the PMDA. Methods We collected data on the antineoplastic drugs approved by the PMDA in fiscal years 2004–2019. We extracted the approval review pathways and the pivotal clinical trial designs from the PMDA review reports, and analyzed them to identify patterns. Results In total, 387 indications in oncology were approved by the PMDA in fiscal years 2004–2019, or 365 indications excluding multiple regulatory pathways. The number of approved indications generally increased year on year (p < 0.001). The largest number of approved indications was under the Orphan Drug Designation (31%, 114/365) and this continues to increase (p < 0.001). In the 288 indications for which clinical trial data were submitted for review, the pivotal clinical trial designs changed significantly (p < 0.001) after the guideline on clinical evaluation for antineoplastic drugs was revised in 2006. Conclusion The number of indications in oncology approved by the PMDA has been increasing over the past 16 years, alongside changes in regulatory pathways. The 2006 guideline on clinical evaluation had a particular impact on pivotal clinical trial designs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Japanese Pharmaceuticals and Medical Devices Agency (PMDA) [1] was established in 2004. Since then, several pieces of legislation, regulations and guidelines have affected the regulatory approval pathways of antineoplastic drugs. These aimed to diversify and speed up the approval review process for domestic drugs. However, no study has yet examined the effects of these measures.
The approval of pharmaceuticals in Japan began with the Pharmaceutical Affairs Law in 1960 [2] and associated regulations in 1961 [3]. These established normal, priority and expedited review pathways, and the orphan drug designation system. In 1993, the Pharmaceutical Affairs Law was revised to facilitate the priority review pathway and orphan drug designation system [4]. In 1999, a notice on the handling of ethical drugs for off-label use was issued [5]. In 2004, a notice on the approval review of drugs evaluated by the Review Committee on Combination Therapy with Antineoplastic Drugs was issued [6]. In 2010, a notice on the prior evaluation of public knowledge was issued [7]. In 2015, a notice on the definition of orphan diseases was issued, which extended the orphan disease definition to cover designated intractable diseases regardless of the number of patients, as well as diseases with less than 50,000 patients in Japan [8]. These changes have diversified the approval review process in Japan. In 2014, the SAKIGAKE designation system was launched on a trial basis to shorten the review period (Table 1, Supplementary Table 1) [9]. In 2017, a conditional early approval system was launched on a trial basis to shorten the development period of new drugs by allowing approval through exploratory clinical trials [10]. These systems were institutionalized in 2019 as part of the amendment to the Pharmaceutical and Medical Devices Law [11, 12]. The PMDA has therefore speeded up the approval review process. To ensure the safety of drugs, the PMDA issued notices on “Risk Management Plan Guidance” in 2012, “Publication of Risk Management Plans” in 2013, and “Risk Management Plan templates and instructions” in 2012 and 2017 [13]. These changes have enhanced and strengthened post-marketing safety measures, by combining risk management with clinical development, review, and post-marketing into a single document. The guideline on clinical evaluation methods for antineoplastic drugs was established in 1991 [14, 15]. This guideline provided recommendations on overall response rate as primary endpoints for pivotal cancer clinical trials for drug efficacy evaluation. The guideline was revised in April 2006 and changed the recommendation of primary endpoints of pivotal trials for drug efficacy evaluation from overall response rate to overall survival [14, 15]. These guidelines have been used to examine primary endpoints and primary analyses of clinical trials for efficacy evaluation of antineoplastic drugs in Japan.
In the US, the safety requirement for new drug products was established in 1938. Its amendment to require that a drug is shown to be effective through substantial evidence was enacted in 1962 (the Federal Food, Drug and Cosmetic Act) [16]. Accelerated approval regulations and priority review designation were established in 1992 (21 Code of Federal Regulations [CFR] part 314, subpart H, 21 CFR part 601 subpart E, and Prescription Drug User Fee Act) for patients with serious or life-threatening diseases [16, 17] (Table 1, Supplementary Table 1). A new drug product can be approved based on an effect on a surrogate endpoint [16]. Once accelerated approval drugs are granted marketing authorization, the US Food and Drug Administration (FDA) requires the sponsors to complete confirmatory trials to describe and verify clinical efficacy [18]. Orphan drug legislation was also enacted in 1992 (21 CFR part 316, subpart C) [19]. A fast-track regulation was established in 1997, and breakthrough therapy regulation in 2012 [17]. Guidance was issued in 2014 for fast track, breakthrough therapy, and priority review designation for serious illnesses, and in 2018 on a real-time oncology review pilot program. These accelerated the regulatory process for antineoplastic drug development in the era of precision medicine, ahead of Japan (Table 1) [17, 20, 21]. The FDA approved 65 novel oncology drugs, and 71 initial indications between 2011 and 2017 [22]. The approvals were based on overall survival data for 15 of the 71 (21%) indications and surrogate endpoints data for 54 of the 71 (76%) indications [22].
In the EU, the European Medicines Agency (EMA) was set up in 1995 to harmonize the work of existing national regulatory bodies. The regulations on accelerated assessment for drugs with a major public health interest and therapeutic innovation, the conditional marketing authorization for life-threatening diseases, and the marketing authorization under exceptional circumstances were all established in 2004 (No 726/2004) [23,24,25] (Table 1, Supplementary Table 1). The orphan designation regulation was enacted in 2000 (No 141/2000) [26]. In 2016, the EMA launched a scheme called PRIME to increase support for the development of medicines targeting an unmet medical need [27].
Overall Survival is a true endpoint in oncology clinical trials and can be used to demonstrate direct clinical benefit to support regulatory approval by the PMDA, FDA, and EMA [28,29,30]. Overall response rate is a surrogate endpoint that assesses the efficacy of a drug by measuring the percentage of complete or partial responses and can be used to support accelerated approval [28,29,30].
This study aimed to examine the impact of the regulatory approval pathways on the antineoplastic drugs approved by PMDA over the past 16 years (fiscal years 2004–2019), analyze the impact on clinical trial designs affecting efficacy evaluation, and assess the impact of particular legislative measures.
Materials and methods
Data source
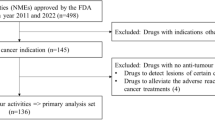
This study included all antineoplastic drugs approved in Japan by the PMDA from April 1, 2004 to March 31, 2020, based on scientific reviews of marketing authorization applications evaluating efficacy and safety of clinical data. All data were obtained from the PMDA website [31]. Analysis included approvals for both initial and supplemental new drug applications for new drug indications and new formulations of drugs. When several indications were approved in one application, each was treated as a separate application.
Data extraction
We collected data about multiple indications for antineoplastic drugs used in the treatment of multiple malignant tumors. For each indication, we extracted information about regulatory approval pathways and pivotal clinical trial designs for drug efficacy evaluation from the PMDA’s review reports, which are available from the PMDA website [32]. Data used for analysis were as follows: tumor types, regulatory pathways for marketing authorization (e.g. normal review, priority review, orphan drug designation, SAKIGAKE designation, conditional early approval, expedited review, review for off-label use, review for drug combination therapy, and prior evaluation of public knowledge), fiscal year approved in Japan, primary endpoint (e.g. overall survival, overall response rate, progression-free survival, disease-free survival, event-free survival, invasive disease-free survival, metastasis-free survival, relapse-free survival, time to progression, complete response, molecular response, cytogenetic response, hematologic response, frequency of other events, duration of other events, and quality of life), phase (e.g. I, I/II, I/III, II, II/III, and III), randomization (e.g. randomized controlled trial and non-randomized controlled trial), comparator (e.g. placebo, active, and none), blindness (e.g. blind and open), clinical trial location (e.g. global, overseas, and local), and sample size. When there were multiple pivotal studies for a single new drug indication, we selected the study with the largest sample size for primary efficacy analysis. When there were multiple primary endpoints in one pivotal trial, we selected one endpoint using the hierarchy: overall survival; progression-free survival; overall response rate; molecular response; cytogenetic response; hematologic response.
Statistical analysis
We performed a Jonckheere-Terpstra trend test to analyze the biennial number of all approved indications over the past 16 years, the biennial number of approved indications under the orphan drug designation or normal review system, and the biennial number of approved indications under prior evaluation of public knowledge. We used a Fisher’s exact test to compare clinical trial designs between fiscal year 2004–2005 and fiscal year 2006–2019 by each of the following items: primary endpoint, phase, randomization, comparator, blindness, location, and sample size. All reported p values used two-sided tests, and those less than 0.05 were considered statistically significant. All statistical analyses used SAS software (version 9.4, SAS Inc.).
Results
Characteristics of the antineoplastic drugs approved in Japan
In total, 387 indications for 339 antineoplastic drugs were approved in Japan by the PMDA between April 1, 2004 and March 31, 2020. Of these 387 indications, 101 (26%) were classified as initial new drug applications, and 288 (74%) as supplemental. Fourteen tumor types accounted for 286 (74%) indications of drugs: malignant lymphoma (12%, 45/387), breast cancer (9%, 36/387), lung cancer (9%, 34/387), leukemia (7%, 29/387), multiple myeloma (7%, 28/387), melanoma (5%, 18/387), colorectal cancer (4%, 17/387), hematological tumor (3%, 13/387), renal cancer (3%, 13/387), unspecified tumor (3%, 12/387), gastric cancer (3%, 11/387), pancreatic cancer (3%, 10/387), malignant solid tumor (3%, 10/387), and prostate cancer (3%, 10/387).
Analysis of biennial changes in the number of indications approved by the PMDA
Figure 1 shows the biennial number of the 365 indications for each regulatory approval pathway, excluding 22 multiple pathways, for anticancer drugs approved by the PMDA from fiscal year 2004 to 2019. The number of approved indications for all regulatory approval pathways has generally increased over the past 16 years (p < 0.001). The largest number of approved indications was under the Orphan Drug Scheme (31%, 114/365), and the number has generally increased over time (p < 0.001). The second largest group used the normal review process (30%, 108/365) and this figure also increased over time (p < 0.001) (Fig. 1). A notice on the approval review of drugs evaluated by the Review Committee on Combination Therapy with Antineoplastic Drugs was issued in 2004 (Fig. 2) on the regulatory approval pathways related to expedited review. The indications under this review for drug combination therapy accounted for 62% (31/50) of all approved indications in fiscal year 2004–2005, but there were no approvals after fiscal year 2006 (Fig. 1). In 2010, a notice on the application for prior evaluation of public knowledge was issued (Fig. 2), and the indications approved under this accounted for 30% (14/46) of all approved indications in fiscal year 2010–2011. However, the number has been decreasing since then (p < 0.001) (Fig. 1).

Number of biennial indications in oncology approved by the PMDA in fiscal years 2004–2019 using each of the regulatory pathways

Laws, notices, and guidelines relevant to regulatory approval review of antineoplastic drugs, issued in Japan in fiscal years 2004–2019
Analysis of biennial changes in the characteristics of pivotal clinical trials
Table 2 shows the biennial changes in the characteristics of clinical trials used for efficacy assessment in the PMDA review reports for the 288 indications for which clinical trial data were submitted to the PMDA for review of antineoplastic drugs approved in fiscal years 2004–2019. The characteristics of pivotal clinical trials for efficacy assessment showed statistically significant changes in primary endpoint, phase, randomization, comparator, location, and sample size between fiscal years 2004–2005 and 2006–2019 (all p < 0.001). No significant change was observed in the presence or absence of blindness (p = 0.519). For the primary endpoint, there was an increase in the proportion of overall survival from 0% (0/12) to 32% (88/276), and progression-free survival from 0% (0/12) to 26% (71/276), and a decrease in the proportion of overall response rate from 58% (7/12) to 20% (56/276), and complete response from 17% (2/12) to 2% (5/276). There was a reduction in the proportion of phase II from 83% (10/12) to 21% (59/276) and an increase in the proportion of phase III from 8% (1/12) to 70% (192/276). There was also an increase in the proportion of randomized controlled trial from 17% (2/12) to 74% (205/276). Similarly, there was a reduction in the proportion with no control arm from 83% (10/12) to 27% (74/276) and a corresponding increase in the proportion with active drug arm from 17% (2/12) to 48% (133/276). We also found an increase in the proportion of global from 0% (0/12) to 46% (128/276) and a corresponding decrease in the proportion of local from 75% (9/12) to 17% (47/276). There was an increase in the median sample size from 53 to 418.
The biennial trends in the primary endpoint and phase were shown in the pivotal clinical trials reviewed by the PMDA for efficacy evaluation during fiscal year 2004–2019 (Fig. 3), indicating a major shift before and after 2006. In clinical trial design during 2004–2005, there were more trials using overall response rate as primary endpoint and more Phase II trials. However, clinical trials from 2006 to 2019 were more likely to use overall survival as the primary endpoint and there were more Phase III trials. Use of overall response rate decreased from 58 to 20% and use of overall survival increased from 0 to 32% after 2006 (p = 0.006, 0.021). The proportion of phase II clinical trials decreased from 83 to 21% with a corresponding increase from 8 to 70% for phase III trials after 2006 (both p < 0.001).

Major shifts in primary endpoint and phase before and after 2006 in pivotal clinical trials reviewed by the PMDA for efficacy evaluation
Discussion
In Japan, 387 indications for antineoplastic drugs were approved by the PMDA in fiscal years 2004–2019. The biennial number of approved indications has generally increased over the past 16 years. This is because of advances in science and technology and understanding of cancer, as well as the research and development of drugs with new mechanisms. To respond to this, the PMDA has diversified and accelerated the approval review process using various measures such as issuing notices, expanding the orphan drug designation system, and introducing the SAKIGAKE designation review system and the conditional early approval system (Table 1, Supplementary Table 1). It is therefore not surprising that the number of approved indications increased. The number of indications approved under the orphan drug designation system was particularly high, suggesting that the hurdles to domestic drug development for rare diseases had been lowered by the PMDA’s work to improve the process for these diseases since the Pharmaceutical and Medical Device Act was enacted in 2013, and the guidelines on risk management plans were issued in 2012 and 2017 (Fig. 2).
After 2006, there were major shifts in the characteristics of clinical trials, which were important for the PMDA’s evaluation of efficacy. There were also several changes in legislation at that time. For example, notices on the review of drug combination therapy and the handling of the priority review process were issued in 2004, and the guideline on clinical evaluation methods for antineoplastic drugs was revised in April 2006. The main changes in the characteristics of pivotal clinical trials were shifts in primary endpoint from overall response rate to overall survival, from phase II to phase III, and from non-randomized controlled trial to randomized controlled trial. These changes were in line with the revised guidelines on clinical evaluation of antineoplastic drugs. We therefore infer that the effect on the design of clinical trials was from the guidelines rather than the notices. This change could be seen as positive from the point of evidence-based medicine because it has encouraged the approval of new antineoplastic drugs that prolong overall survival.
There was a further revision of the guidelines on clinical evaluation methods for antineoplastic drugs in March 2021 [33]. The characteristics of pivotal clinical trials for efficacy evaluation may therefore change significantly in the future, as was the case following the 2006 revision. As the development of antineoplastic drugs for rare cancers and rare cancer subtypes progresses, the number of reviews that evaluate efficacy based on overall response rate in single-arm trials without a control arm may increase in the future. It is expected that the number of accelerated approvals using surrogate endpoints such as overall response rate will increase like the US and the EU. We believe that it is necessary to verify the validity of surrogate endpoints to ensure the reliability of efficacy assessment, as in recent research [34]. The possibility of using the Bayesian approach in trials for rare cancers and rare subtypes is also being examined, and both pharmaceutical companies and the PMDA will need to train people in this approach. Biostatistical aspects of single-arm clinical trials using patient registry data as an external control are also being examined. The design of clinical trials for evaluating the efficacy of antineoplastic drugs is therefore expected to change significantly in the future [35].
Traditionally, Japan has issued notifications rather than enacting new laws to address the operational aspects of the existing system (Supplementary Table 1). Recently, however, the trend toward accelerated approval has been growing, and Japan has caught up with the US and EU by creating the SAKIGAKE designation and conditional early approval (Table 1, Supplementary Table 1). Most recently in the US and EU, attention has been focused on whether new drugs launched by accelerated approval can demonstrate efficacy in post-approval clinical trials [18, 22, 30], and the same will probably be true in Japan.
Our study had some limitations. First, we did not consider the appropriateness of the primary endpoints, comparators, treatments, randomization, blindness, and geographic location of clinical trials. We also did not assess whether surrogate endpoints reflected clinical benefit in the specific indication approved by the PMDA. Instead, we assumed that the clinical trial designs in the PMDA review reports were appropriate. Further studies might be necessary to confirm that a surrogate endpoint reflects clinical benefit and is appropriate as a primary endpoint of a pivotal trial for each indication. Second, we did not take into account studies with negative results for the drug indications if they were not reported in the PMDA review reports. Our findings might therefore not reflect the overall evidence about the authorized indications of antineoplastic drugs. Third, we did not have time to see any change after the recent revision of the guidelines for clinical evaluation of antineoplastic agents. Further studies might be necessary to examine the use of surrogate endpoints in a few years.
In conclusion, we analyzed changes in the regulatory approval system for antineoplastic drugs in Japan over the past 16 years. We found that the number of approved indications has increased, and that the approval review process has become faster and more diverse. We also analyzed changes in the characteristics of pivotal clinical trials for efficacy evaluation of antineoplastic drugs approved by the PMDA over the period, and examined the impact of legislation, notices, and guidelines. We found that the 2006 revision of the guideline on clinical evaluation methods for antineoplastic drugs has had the strongest effect on clinical trial designs. The guideline revisions in March 2021 may therefore lead to further significant changes in pivotal clinical trial designs for efficacy assessment in the future.
Availability of data and material
All data are publicly available from the sources in references.
Code availability
Not applicable.
References
PMDA (2005) https://www.pmda.go.jp/english/about-pmda/outline/0002.html. Accessed 22 Mar 2021
Ministry of Internal Affairs and Communications (MIC) (1960) https://elaws.e-gov.go.jp/document?lawid=335AC0000000145 (Japanese). Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (1961) https://www.mhlw.go.jp/web/t_doc?dataId=81006000&dataType=0&pageNo=1 (Japanese). Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (1993) https://www.mhlw.go.jp/web/t_doc?dataId=00ta6846&dataType=1&pageNo=1 (Japanese). Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (1999) https://www.mhlw.go.jp/shingi/2005/01/dl/s0124-9h1.pdf (Japanese). Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (2004) https://www.hospital.or.jp/pdf/14_20040709_01.pdf (Japanese). Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (2010) Ministry of Health, Labour and Welfare (MHLW).https://www.hospital.or.jp/pdf/18_20100830_01.pdf (Japanese). Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (2016) https://www.nibiohn.go.jp/nibio/part/promote/files/48f09eb927520a338f402e9150d960be0969b81e.pdf (Japanese). Accessed 22 Mar 2021
PMDA (2014) https://www.pmda.go.jp/english/review-services/reviews/advanced-efforts/0001.html. Accessed 22 Mar 2021
PMDA (2017) https://www.pmda.go.jp/files/000227089.pdf. Accessed 22 Mar 2021
PMDA (2020) https://www.pmda.go.jp/review-services/drug-reviews/0002.html (Japanese). Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (2020) https://www.mhlw.go.jp/content/11120000/000666236.pdf (Japanese). Accessed 22 Mar 2021
PMDA (2017) https://www.pmda.go.jp/english/safety/info-services/drugs/rmp/0001.html. Accessed 22 Mar 2021
Ministry of Health, Labour and Welfare (MHLW) (2005) https://www.pmda.go.jp/files/000206740.pdf (Japanese). Accessed 22 Mar 2021
Maeda H, Kurokawa T (2015) Acceptance of Surrogate End Points in Clinical Trials Supporting Approval of Drugs for Cancer Treatment by the Japanese Regulatory Agency. Ann Oncol 26(1):211–216
McKee AE, Farrell AT, Pazdur R, Woodcock J (2010) The Role of the U.S. Food and Drug Administration Review Process: Clinical Trial Endpoints in Oncology. Oncologist 15 Suppl 1:13–18
FDA (2014) https://www.fda.gov/files/drugs/published/Expedited-Programs-for-Serious-Conditions-Drugs-and-Biologics.pdf. Accessed 22 Mar 2021
Naci H, Smalley KR, Kesselheim AS (2017) Characteristics of Preapproval and Postapproval Studies for Drugs Granted Accelerated Approval by the US Food and Drug Administration. JAMA 318(7):626–636
FDA (1992) https://www.ecfr.gov/cgi-bin/retrieveECFR?gp=&SID=718f6fcbc20f2755bd1f5a980eb5eecd&mc=true&n=sp21.5.316.c&r=SUBPART&ty=HTML#se21.5.316_120. Accessed 22 Mar 2021
FDA (2018) https://www.fda.gov/drugs/real-time-review-drug-applications-now-reality-september-20-2018-issue. Accessed 22 Mar 2021
Obama B (2015) Video and transcript: President Obama: Speech on the Precision Medicine Initiative. Available at: http://www.shallownation.com/2015/01/30/president-obama-speech-precision-medicine-initiative-jan-30-2015-white-house/#sthash.Elu9WMPm.dpuf. Accessed 22 Mar 2021
Zettler M, Basch E, Nabhan C (2019) Surrogate End Points and Patient-Reported Outcomes for Novel Oncology Drugs Approved Between 2011 and 2017. JAMA Oncol 5(9):1358–1359
EMA (2004) https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/accelerated-assessment. Accessed 22 Mar 2021
EMA (2004) https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/conditional-marketing-authorisation. Accessed 22 Mar 2021
EMA (2004) https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/pre-authorisation-guidance. Accessed 22 Mar 2021
EMA (2000) https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation/legal-framework-orphan-designation. Accessed 22 Mar 2021
EMA (2016) https://www.ema.europa.eu/en/news/launch-prime-paving-way-promising-medicines-patients. Accessed 22 Mar 2021
Origasa H, Kumagai N, Sumi S (2009) Using and Validating Surrogate Endpoints Toward Accelerated Approvals. Jpn Pharmacol Ther 37:633–639
FDA (2018) https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics. Accessed 22 Mar 2021
Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A (2017) Availability of Evidence of Benefits on Overall Survival and Quality of Life of Cancer Drugs Approved by European Medicines Agency: Retrospective Cohort Study of Drug Approvals 2009–13. BMJ 4(359):j4530
PMDA (2021) https://www.pmda.go.jp/review-services/drug-reviews/review-information/p-drugs/0010.html (Japanese). Accessed 7 Apr 2021
PMDA (2021) https://www.pmda.go.jp/PmdaSearch/iyakuSearch/ (Japanese). Accessed 7 Apr 2021
Ministry of Health, Labour and Welfare (MHLW) (2021) https://public-comment.e-gov.go.jp/servlet/PcmFileDownload?seqNo=0000216748 (Japanese). Accessed 7 Apr 2021
Hirai T, Nemoto A, Ito Y, Matsuura M (2020) Meta-analyses on Progression-Free Survival as a Surrogate Endpoint for Overall Survival in Triple-Negative Breast Cancer. Breast Cancer Res Treat 181(1):189–198
National Cancer Center Japan (2020) https://www.ncc.go.jp/jp/cras/biostatistics/Symposium/symposium.html (Japanese). Accessed 7 Apr 2021
Japan Pharmaceutical Manufacturers Association (2020) http://www.jpma.or.jp/opir/news/061/pdf/no61_p59.pdf (Japanese). Accessed 7 Apr 2021
Acknowledgements
We thank Melissa Leffler, MBA, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Funding
This work was not funded.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Data collection, refinement, and analysis were performed by TH, data collection and refinement were confirmed by AS and TY and data analysis was confirmed by AS, TY, and MM. The first draft of the manuscript was written by TH and all authors approved the final manuscript.
Corresponding author
Ethics declarations
Research involving human participants and/or animals
Not applicable. This article does not discuss any studies with human participants or animals.
Informed consent
Not applicable. This article does not discuss any studies with human participants.
Conflicts of interest
Takehiro Hirai is an employee of Daiichi Sankyo RD Novare. The other authors have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hirai, T., Suzuki, A., Yamori, T. et al. Reforms of regulatory pathways for approval of new antineoplastic drugs in Japan from 2004 to 2019 and accompanying changes in pivotal clinical trial designs. Invest New Drugs 40, 142–150 (2022). https://doi.org/10.1007/s10637-021-01165-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-021-01165-8