
Summary
Neuroblastoma (NB) is the most common extracranial solid tumor in children. Under various treatments, some patients still have a poor prognosis. Hence, it is necessary to find new valid targets for NB therapy. In this study, a comprehensive bioinformatic analysis was used to identify differentially expressed genes (DEGs) between NB and control cells, and to select hub genes associated with NB. GSE66586 and GSE78061 datasets were downloaded from the Gene Expression Omnibus (GEO) database and DEGs were selected. Then, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were applied to the selected DEGs. The STRING database and Cytoscape software were used to construct protein-protein interaction (PPI) networks and perform modular analysis of the DEGs. The R2 database was used for prognostic analysis. We identified a total of 238 DEGs from two microarray databases. GO enrichment analysis shows that these DEGs are mainly concentrated in the regulation of cell growth, cell migration, cell fate determination, and cell maturation. KEGG pathway analysis showed that these DEGs are mainly involved in focal adhesion, the TNF signaling pathway, cancer-related pathways, and signaling pathways regulating stem cell pluripotency. We identified the 15 most closely related DEGs from the PPI network, and performed R2 database prognostic analysis to select five hub genes – CTGF, EDN1, GATA2, LOX, and SERPINE1. This study distinguished hub genes and related signaling pathways that can potentially serve as diagnostic indicators and therapeutic biomarkers for NB, thereby improving understanding of the molecular mechanisms involved in NB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in children and an important cause of childhood cancer deaths. It originates from the neural crest progenitor and mostly occurs in the abdomen, particularly the adrenal gland [1]. Although the incidence rate accounts for only 8% of all childhood cancers, the mortality rate is 15% of the total [2, 3]. The clinical manifestations of patients with NB are diverse. Some patients experience spontaneous degeneration or differentiation into benign ganglioneuroma, whereas others remain affected by tumor hazards after adopting intensive treatment strategies [4, 5]. According to the clinical manifestations and biological characteristics of tumors, including age, disease grade, MYCN expansion or not, and histopathological manifestations, NB patients can be divided into low-, moderate-, and high-risk groups [2, 6]. Patients in the low- and moderate-risk groups responded better to surgery and chemotherapy, and their long-term survival rate was over 90% [7]. Patients in the high-risk group often have extensive metastatic lesions. Even with intense chemotherapy combined with surgery, radiotherapy, and autologous bone marrow stem cell transplantation, their long-term survival rate is still less than 50% [8,9,10]. Improving the cure rate and long-term survival rate of NB patients in the high-risk group is key to improving the overall prognosis, and an urgent problem that needs to be solved in basic research and clinical treatment. Therefore, it is necessary to find new valid targets for NB diagnosis and treatment.
Studies have shown that many biochemical molecular markers are related to tumor occurrence and development and can be used for early tumor screening [11]. However, many markers are highly expressed in various types of tumors and do not have excellent specificity [12]. Therefore, it is necessary to further explore new specific diagnostic NB markers as an auxiliary detection scheme for early diagnosis. Currently, microarray technology and bioinformatic analysis have become a promising and useful tool for screening significant genetic or epigenetic variations that occur during carcinogenesis and determining cancer diagnosis and prognosis [13]. Gene Expression Omnibus (GEO) is an international public repository for the archival and free distribution of microarrays, next-generation sequencing, and other forms of high-throughput functional genomic data [14, 15]. Researchers can obtain publicly available cancer data from around the world, providing opportunities for mining of cancer gene expression profiles [15] and laying a foundation for improving the early diagnosis, treatment, and prevention of various cancers.
In this study, we downloaded two NB chip datasets, GSE66586 and GSE78061, from the GEO database [16, 17]. Differentially expressed genes (DEGs) were screened by comparing gene expression between NB and control cells. A protein-protein interaction (PPI) network was constructed and module analysis of DEGs was performed through the STRING database and Cytoscape software. Then, using gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis in the DAVID database, functional annotation and signal pathway analysis were performed on DEGs, and survival analysis was performed to confirm the importance of prognosis. Finally, CTGF, EDN1, GATA2, LOX, and SERPINE1 are the hub genes. These findings may provide insights into the occurrence and development of NB, as well as potential therapeutic targets for future research.
Materials and methods
Microarray data
Two gene expression datasets (GSE66586, GSE78061) were obtained from the NCBI Gene Expression Synthesis (GEO) database, which can be obtained from http://www.ncbi.nlm.nih.gov/geo/ [18]. The GSE66586 array data was submitted by Gu L. et al., including eight NB and two control cells [17]. The GSE78061 dataset was submitted by Cole KA. et al., and consisted of 25 NB and four control cells [16]. Both datasets are based on the GPL6244 platform (Affymetrix Human Gene 1.0 ST Array; Agilent Technologies, Palo Alto, California, USA).
Microarray data processing
The Affy package in R (http://cran.r-project.org/) was used to perform a robust multi-array average (RMA) algorithm, the original array data was converted into expression values, and background correction, normalization, and probe summary were performed [19, 20]. The paired T-test of the limma package based on R was used to analyze the DEGs between NB and control cells [21, 22]. Adjust P value (AdjP-value) < 0.01 and | log2FC | > 2 were considered as the critical values for DEGs screening.
Integration of microarray data
The list of DEGs obtained from the two microarray datasets was saved as a CSV file through limma packet analysis [21]. We downloaded the robust rank aggregation (RRA) software package and used R to run the instruction code [23]. The gene list was up- or downregulated and the two chips were used for subsequent analysis. The RRA method can be publicly used in the comprehensive R package.
Enrichment analysis of DEGs with GO and KEGG
The GO (http://www.geneontology.org) database can provide a functional classification for genomic data, including biological process (BP), cellular component (CC), and molecular function (MF) [24]. Hence, GO analysis is a widely used gene and gene product annotation tool. The “Kyoto Encyclopedia of Genes and Genomes” (KEGG, http://www.genome.ad.jp/kegg/) database is a networked website designed to analyze, explain, and visualize gene functions [25, 26]. DAVID (http://david.abcc.ncifcrf.gov/) is an annotation, visualization, and comprehensive discovery database, and an online tool for gene function classification, useful for assessing the biological function of genes [27]. In this study, GO enrichment analysis and KEGG pathway analysis were performed using the DAVID website to study the function of DEGs. Values with P < 0.05 were deemed statistically significant.
PPI network construction and analysis
STRING (version: 11.0, https://string-db.org) is a search tool for identifying interacting genes and proteins, and importing DEGs into a database to construct a PPI network, which shows physical and functional interactions [26]. In this study, protein pairs with a total score > 0.4 were selected for PPI network construction. Additionally, Cytoscape software (version 3.6.1) was used to calculate the node degree through the Network Analyzer application, and draw PPI networks with different colors and sizes to show the adjustment (up or down) and node degree [28]. Twelve methods in cytoHubba (Betweenness, BottleNeck, Closeness, Clustering Coefficient, Degree, DMNC, EcCentricity, EPC, MCC, MNC, Radiality, and Stress) were used to rank and evaluate the hub genes, and to finally generate the hub gene network; DAVID was used for hub gene enrichment analysis of GO, and KEGG further explained the reliability of the results.
Hub gene survival analysis
The R2 platform (http://r2platform.com) is a genomic analysis and visualization platform that provides a biologist-friendly interface for high-throughput data analysis. It was developed in the Netherlands AMC Cancer Genomics Department, where it remains the main entry point for all types of high-throughput data. The R2 platform consists of two parts: a publicly accessible database that stores data, coupled with a web interface that provides a set of tools and visualizations to mine the database. In this study, the hub DEGs were selected, and the survival analysis of gene expression in related tumors were performed through the R2 database to determine the relationship between their expression in NB and patient prognoses. Bonfferoni P Value (Bonf P) < 0.05 was regarded as the critical point with statistical significance.
Result
Microarray data information and identification of DEGs
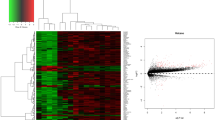
In order to identify DEGs, we performed background correction, and normalization of the NB expression microarray datasets GDS66586 and GSE78061. When filtering the GDS66586 dataset through the limma software package in R (AdjP-value <0.01 and | log2FC | > 2), 778 DEGs were obtained, including 355 upregulated and 423 downregulated DEGs. Besides, 846 DEGs were screened from the GSE78061 dataset, including 500 upregulated and 346 downregulated DEGs. The differential expression of multiple genes in both sample datasets from each of the two microarrays is shown in Fig. 1a and b. In addition, the cluster heatmap of the top 100 DEGs is shown in Fig. 1c and d.

Volcano plot distribution of DEGs and heatmap of the top 100 DEGs between the two datasets. The volcano plot of (a) GSE66586 and (b) GSE78061. The blue points indicate the screened downregulated DEGs, red points indicate the screened upregulated DEGs, and gray points indicate genes with no significant differences; Heatmap for top 100 DEGs of (c) GSE66586 and (d) GSE78061. From red to blue, the expression level of the gene in the sample gradually decreases. All DEGs are screened based on an Adjust P value <0.01 and | fold change | > 2. (DEGs, differentially expressed genes)
Identification of DEGs using integrated bioinformatics
In order to identify overlapped DEGs, we used the limma software package to analyze the two NB gene expression microarray datasets, classified them according to logarithmic change values, and then conducted an RRA analysis (Adjust P value <0.01), which is based on the assumption that each gene in each experiment is randomly ordered. If a gene is ranked higher in all experiments, the smaller the Adjust P value, the higher the likelihood of differential gene expression. RRA analysis showed 238 overlapping DEGs (Table 1), including 151 upregulated and 87 downregulated DEGs (Fig. 2).

Identification of overlapped DEGs. Venn diagram of (a) 151 overlapped upregulated DEGs and (b) 87 overlapped downregulated DEGs between GSE66586 and GSE78061. (DEGs, differentially expressed genes)
GO and KEGG enrichment analysis of overlapped DEGs
In order to understand the molecular functions and pathways involving DEGs, we conducted a functional enrichment analysis. GO-based BP analysis showed that upregulated DEGs were significantly enriched in cell adhesion, extracellular matrix organization, angiogenesis, regulation of cell growth, and cell migration (Fig. 3a), while downregulated DEGs were significantly enriched in negative regulation of transcription, neuron migration, cell fate determination, axon guidance, and cell maturation (Fig. 4a). GO analysis of CC showed that upregulated DEGs were significantly enriched in the plasma membrane, extracellular exosome, extracellular region, extracellular space, and integral component of plasma membrane (Fig. 3b), while downregulated DEGs were mainly involved in the plasma membrane, Golgi apparatus, neuron projection, cell junction, and neuronal cell body (Fig. 4b). Regarding MF, upregulated DEGs were significantly enriched in protein binding, transcription factor activity, RNA polymerase II regulatory region DNA binding, transcriptional activator activity, and RNA polymerase II transcription factor binding (Fig. 3c), while downregulated DEGs were mainly involved in calcium ion binding, actin binding, heparin binding, actin filament binding, and collagen binding (Fig. 4c). In addition, KEGG analysis showed that upregulated DEGs were significantly enriched in focal adhesion, the TNF signaling pathway, and arrhythmogenic right ventricular cardiomyopathy (Fig. 5a), while downregulated DEGs were mainly involved in cholinergic synapse, dopaminergic synapse, morphine addiction, cancer pathways, and signaling pathways regulating stem cell pluripotency (Fig. 5b).

Functional enrichment analysis of upregulated differentially expressed genes (DEGs). Analysis of (a) biological process, (b) cellular component, and (c) molecular function

Functional enrichment analysis of downregulated differentially expressed genes (DEGs). Analysis of (a) biological process, (b) cellular component, and (c) molecular function

Pathway enrichment analysis of DEGs. KEGG pathway analysis of (a) upregulated and (b) downregulated DEGs. (DEGs, differentially expressed genes; KEGG: Kyoto Encyclopedia of Genes and Genomes)
PPI network construction, module analysis, and hub gene determination
In order to study the protein-protein interactions of DEGs, we used STRING network-based protein interaction analysis to generate a PPI network from 238 DEGs overlapped in two datasets (Fig. 6). Following further analysis in Cytoscape, the top 100 DEGs were selected by 12 modules in cytoHubba and intersected. A total of 48 overlapped DEGs were identified and visualized (Fig. 7a). The 48 overlapped DEGs were sorted according to their degree scores, and the top 15 DEGs with the highest scores, namely ACTA2, COL4A1, LOX, CTGF, FBN1, SERPINE1, FSTL1, GATA3, GATA2, TAGLN, ISL1, HAND2, GJA1, MMP14, and EDN1, were selected (Table 2) and visualized (Fig. 7b).

Protein-Protein Interaction (PPI) network of differentially expressed genes (DEGs) constructed in STRING

PPI network of DEGs constructed in Cytoscape. PPI network of (a) overlapped DEGs, filtered out by 12 modules of cytoHubba in Cytoscape, and (b) the top 15 overlapped hub DEGs. Red points represent upregulated DEGs, and green points represent downregulated DEGs. (DEGs, differentially expressed genes; PPI, protein-protein interaction)
Hub gene survival analysis
In order to study the correlation between DEG expression and NB patient prognosis, we further analyzed data for 649 samples through R2 database. Finally, we selected the prognosis of CTGF, EDN1, GATA2, LOX, SERPINE1, and NB patients in these 15 DEGs. The closely related DEGs serve as hub genes. Among them, the overall survival rates of patients with low CTGF and LOX expression and high EDN1, GATA2, and SERPINE1 expression in NB were high (Figs. 8 and 9a-e). Subsequent GO and KEGG enrichment analyses showed that hub genes were mainly enriched in intracellular signal transduction, cell-cell signaling, protein binding, HIF-1 signaling pathway, and Hippo signaling pathway (Table 3), and the visualization results are shown in Fig. 8. Bonf p < 0.05 was regarded as the critical point with statistical significance.

Distribution of hub DEGs in NB for GO enrichment. (DEGs, differentially expressed genes; NB, Neuroblastoma; GO, gene ontology)

Survival Analysis of hub differentially expressed genes (DEGs). Survival Analysis of (a) CTGF, (b) EDN1, (c) GATA2, (d) LOX, and (e) SERPINE1 in NB. Bonf p < 0.05 was regarded as the critical point with statistical significance
Discussion
NB is a significant cause of childhood death, and early diagnosis and treatment are essential to prolonging the survival time of patients [29]. Therefore, it is necessary to further explore the predictive indicators and treatment goals of NB. With the development of bioinformatics, DNA microarrays are increasingly used to explore the early diagnosis, treatment, and prognosis of cancer [30]. This study aimed to identify DEGs between NB and control cells to further understand the pathogenesis of NB and potentially provide diagnostic biomarkers and therapeutic targets.
According to reports, studies using multiple cohorts tend to have lower false positive and false negative rates than single cohort studies [31]. However, due to factors, such as batch effects and biological differences, multiple microarrays from different platforms may obscure and confuse the real situation [32]. In order to improve the reliability of DEG identification, we selected two microarray datasets from the same platform, then identified a total of 704 upregulated and 682 downregulated DEGs. Among them, 151 upregulated and 87 downregulated DEGs were significantly expressed in both datasets. To further define the role of these DEGs in NB, we conducted a series of bioinformatic and prognostic analyses of these DEGs.
GO enrichment analysis showed the upregulation of DEGs mainly involved in BP, such as cell adhesion, regulation of cell growth, and cell migration. In contrast, DEGs mainly involved in BP, such as negative regulation of transcription, cell fate determination, and cell maturation were downregulated. Studies have shown that the reduction in cell adhesion and the change in cell migration ability are critical steps in cancer metastasis, which is consistent with our the results from our analysis [33, 34]. For MF, DEGs mainly involved in protein binding, transcription factor activity, and cadherin binding were upregulated, whereas DEGs mainly enriched in calcium ion binding, actin binding, and heparin binding were downregulated. Villalobos and others pointed out that calmodulin plays an essential role in tumor cell migration, invasion, and metastasis, which supports our findings [35]. CC analysis showed that the upregulation of DEGs was concentrated in the plasma membrane, extracellular exosome, and extracellular region, whereas the downregulation of DEGs was concentrated in the plasma membrane, Golgi apparatus, and cell junction. Some previous studies have shown that the role of extracellular exosome and transcription factor activity in tumor development and progression is consistent with the results of this study [36]. Besides, KEGG enrichment analysis showed the upregulation of DEGs that were significantly enriched in the TNF signaling pathway and the downregulation of DEGs significantly enriched in the cancer pathways, signaling pathways regulating pluripotency of stem cells, and cell adhesion molecules. This is also consistent with the fact that TNF is a cytokine that can directly kill tumor cells and has no apparent cytotoxicity to healthy cells. The activation of the TNF signaling pathway also plays a crucial role in tumor regulation [37, 38].
A PPI network was constructed for the identified DEGs, and critical genes were defined according to degree levels. ACTA2, COL4A1, LOX, CTGF, FBN1, SERPINE1, FSTL1, GATA3, GATA2, TAGLN, ISL1, HAND2, GJA1, MMP14, and EDN1 were determined to have a high degree of network connectivity, combined with gene expression and NB prognosis correlation. We finally identified CTGF, EDN1, GATA2, LOX, and SERPINE1 as hub genes among these 15 genes. Functional enrichment analysis of the hub genes showed that the development of NB is related to angiogenesis, protein binding, the HIF-1 signaling pathway, and the Hippo signaling pathway.
CTGF, also known as CCN2 (Cell Communication Network Factor 2), is a protein-coding gene that encodes proteins that play a role in cell adhesion in many cell types and participates in ERK signaling and the TGF-β pathway [39,40,41,42]. Wang et al. pointed out that in cells overexpressing TAZ, knocking out CTGF with small interfering RNA can inhibit the expression of CTGF induced by TAZ, thereby inhibiting the proliferation and colony formation of NB cells [43]. Although there are relatively few studies on the role of CTGF in NB, this provides some potential value for the study of its role in NB.
EDN1 is a member of the endothelin family and is a protein-coding gene. Abnormal expression of genes may promote tumorigenesis [44]. SERPINE1 is a member of the serpin protease inhibitor (serpin) superfamily, reported to be involved in the Hippo signaling pathway [45]. Although there are no reports related to the research on the role of EDN1 and SERPINE1 in NB, our GO and KEGG enrichment analysis showed that EDN1 and SERPINE1 were involved in the regulation of cell-cell signaling, protein binding, the HIF-1 signaling pathway, and the Hippo signaling pathway. This agrees with previous reports that the HIF-1 and Hippo signaling pathways play a vital role in NB regulation [46,47,48].
GATA2 (GATA binding protein 2) is a protein-coding gene that plays an essential role in regulating the transcription of genes involved in the development and proliferation of hematopoietic and endocrine cell lineages [49]. Hoene et al. Pointed out that changes in the expression levels of GATA2 and its family in NB may be related to the pathogenesis of neuroblastoma [50]. Wei and others pointed out that in the transcriptome sequencing of three tumors including NB, LPAR1, GATA2, and NUFIP1 had high expression levels of mutant alleles, indicating that these mutant genes may have carcinogenic effects [51]. These results indicate that GATA2 may be a potential marker for early cancer detection and prognosis.
LOX is an extracellular copper-dependent amine oxidase, which is involved in the crosslinking of collagen and elastin lysine residues in the extracellular matrix. Its expression level in tumors is related to tumor prognosis [52]. Studies have shown that LOX/COX inhibitors can promote the differentiation of neuroblastoma cells induced by all-trans retinoic acid to a certain extent [53, 54]. Based on the role of LOX in tumors, with further in-depth research, LOX is expected to become a potential target molecule for NB treatment.
In summary, the results from this study suggest that CTGF, EDN1, GATA2, LOX, and SERPINE1 are NB hub genes. GO and KEGG enrichment analysis of these five hub genes further revealed their functions and pathways, and survival analysis found them to be closely associated to the prognosis of NB patients. These genes may become potential markers for improving diagnosis, optimizing chemotherapy, and predicting prognosis for NB, as pathways related to the genes are potential therapeutic targets for NB. We plan to verify the potential functions and pathways of these genes in future research.
Data availability
All data is available under reasonable request.
References
Maris JM, Hogarty MD, Bagatell R, Cohn SL (2007) Neuroblastoma. Lancet 369(9579):2106–2120. https://doi.org/10.1016/S0140-6736(07)60983-0
Fonseka P, Liem M, Ozcitti C, Adda CG, Ang CS, Mathivanan S (2019) Exosomes from N-Myc amplified neuroblastoma cells induce migration and confer chemoresistance to non-N-Myc amplified cells: implications of intra-tumour heterogeneity. J Extracell Vesicles 8(1):1597614. https://doi.org/10.1080/20013078.2019.1597614
Brodeur GM (2003) Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 3(3):203–216. https://doi.org/10.1038/nrc1014
Scheer M, Bork K, Simon F, Nagasundaram M, Horstkorte R, Gnanapragassam VS (2020) Glycation leads to increased Polysialylation and promotes the metastatic potential of Neuroblastoma cells. Cells 9(4). https://doi.org/10.3390/cells9040868
Depuydt P, Boeva V, Hocking TD, Cannoodt R, Ambros IM, Ambros PF, Asgharzadeh S, Attiyeh EF, Combaret V, Defferrari R, Fischer M, Hero B, Hogarty MD, Irwin MS, Koster J, Kreissman S, Ladenstein R, Lapouble E, Laureys G, London WB, Mazzocco K, Nakagawara A, Noguera R, Ohira M, Park JR, Potschger U, Theissen J, Tonini GP, Valteau-Couanet D, Varesio L, Versteeg R, Speleman F, Maris JM, Schleiermacher G, De Preter K (2018) Genomic amplifications and distal 6q loss: novel markers for poor survival in high-risk Neuroblastoma patients. J Natl Cancer Inst 110(10):1084–1093. https://doi.org/10.1093/jnci/djy022
Koneru B, Lopez G, Farooqi A, Conkrite KL, Nguyen TH, Macha SJ, Modi A, Rokita JL, Urias E, Hindle A, Davidson H, McCoy K, Nance J, Yazdani V, Irwin MS, Yang S, Wheeler DA, Maris JM, Diskin SJ, Reynolds CP (2020) Telomere maintenance mechanisms define clinical outcome in high-risk neuroblastoma. Cancer Res 80:2663–2675. https://doi.org/10.1158/0008-5472.CAN-19-3068
Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, Nakagawara A, Berthold F, Schleiermacher G, Park JR, Valteau-Couanet D, Pearson AD, Cohn SL (2015) Advances in risk classification and treatment strategies for Neuroblastoma. J Clin Oncol 33(27):3008–3017. https://doi.org/10.1200/JCO.2014.59.4648
Upton K, Modi A, Patel K, Kendsersky NM, Conkrite KL, Sussman RT, Way GP, Adams RN, Sacks GI, Fortina P, Diskin SJ, Maris JM, Rokita JL (2020) Epigenomic profiling of neuroblastoma cell lines. Sci Data 7(1):116. https://doi.org/10.1038/s41597-020-0458-y
Almstedt E, Elgendy R, Hekmati N, Rosen E, Warn C, Olsen TK, Dyberg C, Doroszko M, Larsson I, Sundstrom A, Arsenian Henriksson M, Pahlman S, Bexell D, Vanlandewijck M, Kogner P, Jornsten R, Krona C, Nelander S (2020) Integrative discovery of treatments for high-risk neuroblastoma. Nat Commun 11(1):71. https://doi.org/10.1038/s41467-019-13817-8
Berthold F, Faldum A, Ernst A, Boos J, Dilloo D, Eggert A, Fischer M, Fruhwald M, Henze G, Klingebiel T, Kratz C, Kremens B, Krug B, Leuschner I, Schmidt M, Schmidt R, Schumacher-Kuckelkorn R, von Schweinitz D, Schilling FH, Theissen J, Volland R, Hero B, Simon T (2020) Extended induction chemotherapy does not improve the outcome for high-risk neuroblastoma patients: results of the randomized open-label GPOH trial NB2004-HR. Ann Oncol 31(3):422–429. https://doi.org/10.1016/j.annonc.2019.11.011
Pstrag N, Ziemnicka K, Bluyssen H, Wesoly J (2018) Thyroid cancers of follicular origin in a genomic light: in-depth overview of common and unique molecular marker candidates. Mol Cancer 17(1):116. https://doi.org/10.1186/s12943-018-0866-1
Gao Y, Huo W, Zhang L, Lian J, Tao W, Song C, Tang J, Shi S, Gao Y (2019) Multiplex measurement of twelve tumor markers using a GMR multi-biomarker immunoassay biosensor. Biosens Bioelectron 123:204–210. https://doi.org/10.1016/j.bios.2018.08.060
Coyle R, Jia J, Mei Y (2016) Polymer microarray technology for stem cell engineering. Acta Biomater 34:60–72. https://doi.org/10.1016/j.actbio.2015.10.030
Toro-Dominguez D, Martorell-Marugan J, Lopez-Dominguez R, Garcia-Moreno A, Gonzalez-Rumayor V, Alarcon-Riquelme ME, Carmona-Saez P (2019) ImaGEO: integrative gene expression meta-analysis from GEO database. Bioinformatics 35(5):880–882. https://doi.org/10.1093/bioinformatics/bty721
Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R (2007) NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic Acids Res 35(Database issue):D760–D765. https://doi.org/10.1093/nar/gkl887
Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, Gagliardi M, Chen L, Martinez D, Li Y, Wood A, Kim S, Parasuraman S, Delach S, Cole KA, Krupa S, Boehm M, Peters M, Caponigro G, Maris JM (2017) Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in Neuroblastoma. Clin Cancer Res 23(7):1785–1796. https://doi.org/10.1158/1078-0432.CCR-16-1131
Gu L, Chu P, Lingeman R, McDaniel H, Kechichian S, Hickey RJ, Liu Z, Yuan YC, Sandoval JA, Fields GB, Malkas LH (2015) The mechanism by which MYCN amplification confers an enhanced sensitivity to a PCNA-derived cell permeable peptide in Neuroblastoma cells. EBioMedicine 2(12):1923–1931. https://doi.org/10.1016/j.ebiom.2015.11.016
Clough E, Barrett T (2016) The gene expression omnibus database. Methods Mol Biol 1418:93–110. https://doi.org/10.1007/978-1-4939-3578-9_5
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4(2):249–264. https://doi.org/10.1093/biostatistics/4.2.249
Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) Affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20(3):307–315. https://doi.org/10.1093/bioinformatics/btg405
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47. https://doi.org/10.1093/nar/gkv007
Diboun I, Wernisch L, Orengo CA, Koltzenburg M (2006) Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics 7:252. https://doi.org/10.1186/1471-2164-7-252
Nie K, Shi L, Wen Y, Pan J, Li P, Zheng Z, Liu F (2019) Identification of hub genes correlated with the pathogenesis and prognosis of gastric cancer via bioinformatics methods. Minerva Med. https://doi.org/10.23736/S0026-4806.19.06166-4
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet 25(1):25–29. https://doi.org/10.1038/75556
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28(1):27–30. https://doi.org/10.1093/nar/28.1.27
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27(1):29–34. https://doi.org/10.1093/nar/27.1.29
Sherman BT, da Huang W, Tan Q, Guo Y, Bour S, Liu D, Stephens R, Baseler MW, Lane HC, Lempicki RA (2007) DAVID knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics 8:426. https://doi.org/10.1186/1471-2105-8-426
Doncheva NT, Morris JH, Gorodkin J, Jensen LJ (2019) Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res 18(2):623–632. https://doi.org/10.1021/acs.jproteome.8b00702
Park JA, Cheung NV (2020) Targets and antibody formats for immunotherapy of Neuroblastoma. J Clin Oncol:JCO1901410. https://doi.org/10.1200/JCO.19.01410
Xia XQ, Jia Z, Porwollik S, Long F, Hoemme C, Ye K, Muller-Tidow C, McClelland M, Wang Y (2010) Evaluating oligonucleotide properties for DNA microarray probe design. Nucleic Acids Res 38(11):e121. https://doi.org/10.1093/nar/gkq039
Pounds S, Morris SW (2003) Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics 19(10):1236–1242. https://doi.org/10.1093/bioinformatics/btg148
Kupfer P, Guthke R, Pohlers D, Huber R, Koczan D, Kinne RW (2012) Batch correction of microarray data substantially improves the identification of genes differentially expressed in rheumatoid arthritis and osteoarthritis. BMC Med Genet 5:23. https://doi.org/10.1186/1755-8794-5-23
Karstens KF, Bellon E, Polonski A, Wolters-Eisfeld G, Melling N, Reeh M, Izbicki JR, Tachezy M (2020) Expression and serum levels of the neural cell adhesion molecule L1-like protein (CHL1) in gastrointestinal stroma tumors (GIST) and its prognostic power. Oncotarget 11(13):1131–1140. https://doi.org/10.18632/oncotarget.27525
Beltran-Anaya FO, Romero-Cordoba S, Rebollar-Vega R, Arrieta O, Bautista-Pina V, Dominguez-Reyes C, Villegas-Carlos F, Tenorio-Torres A, Alfaro-Riuz L, Jimenez-Morales S, Cedro-Tanda A, Rios-Romero M, Reyes-Grajeda JP, Tagliabue E, Iorio MV, Hidalgo-Miranda A (2019) Expression of long non-coding RNA ENSG00000226738 (LncKLHDC7B) is enriched in the immunomodulatory triple-negative breast cancer subtype and its alteration promotes cell migration, invasion, and resistance to cell death. Mol Oncol 13(4):909–927. https://doi.org/10.1002/1878-0261.12446
Villalobo A, Berchtold MW (2020) The role of Calmodulin in tumor cell migration, invasiveness, and metastasis. Int J Mol Sci 21(3). https://doi.org/10.3390/ijms21030765
Haug BH, Hald OH, Utnes P, Roth SA, Lokke C, Flaegstad T, Einvik C (2015) Exosome-like extracellular vesicles from MYCN-amplified Neuroblastoma cells contain oncogenic miRNAs. Anticancer Res 35(5):2521–2530
Ma W, Chen X, Wu X, Li J, Mei C, Jing W, Teng L, Tu H, Jiang X, Wang G, Chen Y, Wang K, Wang H, Wei Y, Liu Z, Yuan Y (2020) Long noncoding RNA SPRY4-IT1 promotes proliferation and metastasis of hepatocellular carcinoma via mediating TNF signaling pathway. J Cell Physiol. https://doi.org/10.1002/jcp.29438
Li M, Ren CX, Zhang JM, Xin XY, Hua T, Wang HB, Wang HB (2018) The effects of miR-195-5p/MMP14 on proliferation and invasion of cervical carcinoma cells through TNF signaling pathway based on bioinformatics analysis of microarray profiling. Cell Physiol Biochem 50(4):1398–1413. https://doi.org/10.1159/000494602
Song Y, Kim JS, Choi EK, Kim J, Kim KM, Seo HR (2017) TGF-beta-independent CTGF induction regulates cell adhesion mediated drug resistance by increasing collagen I in HCC. Oncotarget 8(13):21650–21662. https://doi.org/10.18632/oncotarget.15521
Waddell JM, Evans J, Jabbour HN, Denison FC (2011) CTGF expression is up-regulated by PROK1 in early pregnancy and influences HTR-8/Svneo cell adhesion and network formation. Hum Reprod 26(1):67–75. https://doi.org/10.1093/humrep/deq294
Ball DK, Rachfal AW, Kemper SA, Brigstock DR (2003) The heparin-binding 10 kDa fragment of connective tissue growth factor (CTGF) containing module 4 alone stimulates cell adhesion. J Endocrinol 176(2):R1–R7. https://doi.org/10.1677/joe.0.176r001
Song ZM, Liu F, Chen YM, Liu YJ, Wang XD, Du SY (2019) CTGF-mediated ERK signaling pathway influences the inflammatory factors and intestinal flora in ulcerative colitis. Biomed Pharmacother 111:1429–1437. https://doi.org/10.1016/j.biopha.2018.12.063
Wang M, Liu Y, Zou J, Yang R, Xuan F, Wang Y, Gao N, Cui H (2015) Transcriptional co-activator TAZ sustains proliferation and tumorigenicity of neuroblastoma by targeting CTGF and PDGF-beta. Oncotarget 6(11):9517–9530. https://doi.org/10.18632/oncotarget.3367
Yuan W, Qian M, Li ZX, Zhao CL, Zhao J, Xiao JR (2019) Endothelin-1 activates the notch signaling pathway and promotes tumorigenesis in Giant cell tumor of the spine. Spine (Phila Pa 1976) 44(17):E1000–E1009. https://doi.org/10.1097/BRS.0000000000003044
Basurto L, Sanchez L, Diaz A, Valle M, Robledo A, Martinez-Murillo C (2019) Differences between metabolically healthy and unhealthy obesity in PAI-1 level: fibrinolysis, body size phenotypes and metabolism. Thromb Res 180:110–114. https://doi.org/10.1016/j.thromres.2019.06.013
Tang W, Dong K, Li K, Dong R, Zheng S (2016) MEG3, HCN3 and linc01105 influence the proliferation and apoptosis of neuroblastoma cells via the HIF-1alpha and p53 pathways. Sci Rep 6:36268. https://doi.org/10.1038/srep36268
Chen SJ, Hoffman NE, Shanmughapriya S, Bao L, Keefer K, Conrad K, Merali S, Takahashi Y, Abraham T, Hirschler-Laszkiewicz I, Wang J, Zhang XQ, Song J, Barrero C, Shi Y, Kawasawa YI, Bayerl M, Sun T, Barbour M, Wang HG, Madesh M, Cheung JY, Miller BA (2014) A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2alpha. J Biol Chem 289(52):36284–36302. https://doi.org/10.1074/jbc.M114.620922
Wang Q, Xu Z, An Q, Jiang D, Wang L, Liang B, Li Z (2015) TAZ promotes epithelial to mesenchymal transition via the upregulation of connective tissue growth factor expression in neuroblastoma cells. Mol Med Rep 11(2):982–988. https://doi.org/10.3892/mmr.2014.2818
Mendes-de-Almeida DP, Andrade FG, Borges G, Dos Santos-Bueno FV, Vieira IF, da Rocha L, Mendes-da-Cruz DA, Zancope-Oliveira RM, Calado RT, Pombo-de-Oliveira MS (2019) GATA2 mutation in long stand Mycobacterium kansasii infection, myelodysplasia and MonoMAC syndrome: a case-report. BMC Med Genet 20(1):64. https://doi.org/10.1186/s12881-019-0799-6
Hoene V, Fischer M, Ivanova A, Wallach T, Berthold F, Dame C (2009) GATA factors in human neuroblastoma: distinctive expression patterns in clinical subtypes. Br J Cancer 101(8):1481–1489. https://doi.org/10.1038/sj.bjc.6605276
Wei JS, Johansson P, Chen L, Song YK, Tolman C, Li S, Hurd L, Patidar R, Wen X, Badgett TC, Cheuk AT, Marshall JC, Steeg PS, Vaque Diez JP, Yu Y, Gutkind JS, Khan J (2013) Massively parallel sequencing reveals an accumulation of de novo mutations and an activating mutation of LPAR1 in a patient with metastatic neuroblastoma. PLoS One 8(10):e77731. https://doi.org/10.1371/journal.pone.0077731
Li Q, Zhu CC, Ni B, Zhang ZZ, Jiang SH, Hu LP, Wang X, Zhang XX, Huang PQ, Yang Q, Li J, Gu JR, Xu J, Luo KQ, Zhao G, Zhang ZG (2019) Lysyl oxidase promotes liver metastasis of gastric cancer via facilitating the reciprocal interactions between tumor cells and cancer associated fibroblasts. EBioMedicine 49:157–171. https://doi.org/10.1016/j.ebiom.2019.10.037
Redova M, Chlapek P, Loja T, Zitterbart K, Hermanova M, Sterba J, Veselska R (2010) Influence of LOX/COX inhibitors on cell differentiation induced by all-trans retinoic acid in neuroblastoma cell lines. Int J Mol Med 25(2):271–280
Chlapek P, Redova M, Zitterbart K, Hermanova M, Sterba J, Veselska R (2010) Enhancement of ATRA-induced differentiation of neuroblastoma cells with LOX/COX inhibitors: an expression profiling study. J Exp Clin Cancer Res 29:45. https://doi.org/10.1186/1756-9966-29-45
Acknowledgements
We thank the staff from Medical Research Center of Shengjing Hospital who gave us support throughout the experiments.
Funding
The work was supported by the National Natural Science Foundation of China (No. 81972515, 81,472,359), Key Research and Development Foundation of Liaoning Province (2019JH8/10300024), 2013 Liaoning Climbing Scholar Foundation, and 345 Talent Project of Shengjing Hospital of China Medical University.
Author information
Authors and Affiliations
Contributions
Bo Chen and Peng Ding had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Both contributed equally to the study and are co-first authors; Zhongyan Hua and Xiuni Qin contributed to collection of data; Zhijie Li contributed to the study design, interpretation of the data, the writing of the manuscript, and the submission of the manuscript for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Consent to participate
Not applicable.
Consent for publication
All authors consent to the publication of this study.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, B., Ding, P., Hua, Z. et al. Analysis and identification of novel biomarkers involved in neuroblastoma via integrated bioinformatics. Invest New Drugs 39, 52–65 (2021). https://doi.org/10.1007/s10637-020-00980-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-020-00980-9