
Abstract
Expression of CD20 antigen by the most of transformed B cells is believed to be the driving force for targeting this molecule by using anti-CD20 monoclonal antibodies. While it is true that most lymphoma/leukemia patients can be cured, these regimens are limited by the emergence of treatment resistance. Based on these observations, development of anti-CD20 monoclonal antibodies and combination therapies have been recently proposed, in particular with the aim to optimize the cytotoxic activity. Here we outline a range of new experimental agents concerning the CD20 positive B-cell tumors which provide high benefit from conventional therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
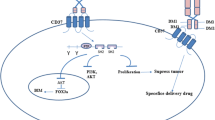
CD20 molecule is a phosphoprotein highly expressed on malignant B cells in non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia (CLL), making these malignancies susceptible to immunotherapeutic strategies [1]. The exact function of CD20 antigen is still unknown, but it is thought to play roles in B-cell activation, differentiation and regulation of calcium influx [2, 3]. Several recent studies have provided interesting clues illustrating the importance of CD20 as an attractive target for immunotherapeutic strategies, including (i) highly expression in over 90 % of neoplastic B cells, but not on hematological stem cells or terminally differentiated plasma cells, allowing clustered opsonization of tumor cells by monoclonal antibodies (mAbs), (ii) tight junction with plasma membrane that contributes to binding of mAbs in close proximity to the cell surface, (iii) lack of physiological ligands, which might interfere with anti-CD20 mAb binding, and (iv) resistance to internalization, shedding or much modulation upon binding by an anti-CD20 antibody [4, 5]. Despite significant therapeutic advances by using anti-CD20 antibodies such as rituximab, most mature B-cell malignancies are also incurable and there is a need for new therapies [6]. In an effort to improve the outcome of CD20 targeted therapy in B-cell neoplasis, many strategies such as antibody engineering approaches, radiotherapy and cell based immunotherapy have been used. Moreover, current clinical trials focus on combination therapies that are even more effective for relapsed or progressive B-Lineage disease. We will review herein the treatment strategies for B-cell malignancies to provide exciting opportunities for new and improved therapeutics agents.
Biology of CD20 antigen
The CD20 antigen belongs to transmembrane protein of the tetraspanin superfamily that is exclusively expressed on the pre-B-cell stage, but is absent on plasma cells and hematopoietic stem cells. The molecular biology of CD20 and its potential as a target for mAb therapy are key elements for its clinical success [7].
Hydropathicity analysis of its sequence predicted three hydrophobic regions consisting: a tetraspan transmembrane molecule with two extracellular loops, a larger one of ~44 amino acids between the third and fourth transmembrane regions, and a much smaller one between the first and second hydrophobic domains as well as an intracellular N and C-terminal regions with multiple consensus sequences for phosphorylation [8]. A better understanding of new epitope determination within a desired part of human CD20 antigen using in silico tools would be useful to develop synthetic peptide vaccines, immunodiagnostic tests and mAbs with different binding sites. Using phage display libraries, Binder et al. showed that rituximab binds a discontinuous epitope within the extracellular segment of CD20, consisting (170) ANPS (173) and (182) YCYSI (185), both strings brought in steric proximity by a disulfide bridge between C (167) and C (183) [9].
Human CD20 is not glycosylated, but multiple isoforms of this antigen resulting from differential phosphorylation states with Mr of 33 kD (most abundant in normal unstimulated canine peripheral B cells) and Mr of 34 to 36 kD (predominant forms seen in malignant B-cell lines) have been identified [10]. Besides phosphorylation, respective palmitoylation of CD20 is required for its immune activity and cell responses to anti-CD20 immunotherapy [11]. Initially, CD20 has been considered unlikely to be shed from the surface of cells, but subsequently it was reported that this antigen is present at abnormal levels in the sera of patients with NHL, both before and after first-line non-mAb containing therapy [12]. Although the exact biological function of CD20 has not yet been clarified, its participation in the B-cell activation, differentiation and regulation of calcium influx has been supposed. More specifically, CD20 expression level in B-cell lines has been reported to play a critical role in calcium influx across the plasma membrane. A recent case report of a patient with CD20 deficiency also suggested a central role in the generation of T-cell-independent immune responses [13, 14]. There is now overwhelming evidence that CD20 is physically and functionally associated to cellular signaling cascades and display multiple interactions with specific B-cell surface markers such as fibroblast growth factor receptor 3 (FGFR3), CD40, B cell receptor (BCR), and complement receptor CD21 [15, 16].
Developing therapeutic strategies for CD20+ B cells
Recombinant antibody engineering has become a popular technology in the treatment of disorders over the last 30 years [17]. The first report of antibody production specific for CD20 antigen (murine anti-CD20 B1) was introduced in 1980 and since then several murine antibodies have been developed to target this molecule. Rituximab, approved in 1997, was the first anti-CD20 chimeric mAb used for treatment of patients with several forms of B-cell–derived neoplasm as CLL, follicular lymphoma (FL), mantle cell lymphoma (MCL) and diffuse large B-cell lymphoma (DLBCL) [18].
Accumulating evidence indicates that some factors, including CD20 cell-surface expression level, the presence of a mutation/deletion in this antigen, CD20 protein phosphorylation rate [19], CD20 modulation/endocytosis [20], Lipid raft composition [21], epigenetic regulation of the CD20 gene, Fcγ Receptor (FcγR) and C1q genes polymorphisms [22, 23], expression of FcγRIIB, complement regulatory proteins (CRP) CD59 and CD55 overexpression [24], Local tumor burden [25], and cell adhesion mediated antibody resistance (CAM-AR) are commonly associated with clinical benefit from rituximab treatment. On the basis of what above reported, scientific efforts are increasingly being focused on developing innovative tailored strategies to improve mAb-driven cytotoxicity. We will provide an overview of these approaches in the next sections.
Depleting neoplastic B cells with novel anti-CD20 mAbs
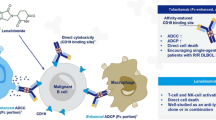
Based on their mode of CD20 binding and primary mechanism of action for killing CD20-positive B-cell neoplasms, anti-CD20 mAbs can be categorized into type I (eg., rituximab, ofatumumab) and type II (eg., tositumumab, GA101) [26–33] (Table 1). Both types of these antibodies are supposed to work mainly by mediating antibody dependent cellular cytotoxicity (ADCC). It is still not clear what characteristics are required for the optimal reagent, but it is generally accepted that Fc-dependent effector mechanisms are crucial for the efficacy of anti-CD20 mAbs. As mentioned above, despite effective antigen targeting capacity, rituximab usually exhibit undesirable side-effects which restrict their applications in treatment of human diseases.
To overcome these drawbacks, effective anti-CD20 antibodies are urgently needed [34–43] (Table 2). Central to these findings are the implications for targeting this antigen by using humanized antibodies. Obinutuzumab (GA101) is a fully humanized glycoengineered type II CD20 antibody which elucidates increased ADCC and a higher degree of apoptosis than rituximab. More importantly, obinutuzumab induces lysosome-dependent cell death in CLL cells receiving activation signals through CD40-CD40 Ligand (CD40L) interactions [44]. The recent efforts to understand the function of obinutuzumab indicate that this agent significantly inhibits cell proliferation and down-regulates PI3K/Akt and NF-κB signaling pathways as well as induces programmed cell death in primary mediastinal large B-cell lymphoma (PMBL) [45]. A phase II, randomized trial of obinutuzumab monotherapy showed significant efficacy, for 1000 mg as well as for 2000 mg, in untreated CLL patients with no unexpected toxicities [46].
Ofatumumab is another fully humanized anti-CD20 mAb that recognizes membrane-proximal small-loop on CD20, demonstrated greatest complement activation as compared with rituximab and obinutuzumab [47].
Ofatumumab in combination with bendamustine has advanced into phase III clinical trials for rituximab-resistant indolent NHL patients (iNHL) [NCT01077518].
Based on published data by Doubek et al., ofatumumab–dexamethasone (O-Dex) regimen exhibited significant clinical benefit for patients with relapsed/refractory CLL, including those with p53 abnormalities [48].
Veltuzumab is a humanized anti-CD20 mAb constructed recombinantly on the framework regions of anti-CD22 mAb epratuzumab could favor ADCC and apoptosis [49]. In a recent report, subcutaneous administration of veltuzumab in 17 patients with refractory iNHL was well tolerated with no serious side effects [50]. A phase I/II trial of veltuzumab and milatuzumab, a humanized anti-CD74 antibody, was conducted in patients with relapsed or refractory B-cell NHL. In regard to safety, immunotherapy-associated grade 3–4 adverse events included anemia, atrial tachycardia, fatigue, hyperglycemia, infusion reactions, leukopenia, lymphopenia, and neutropenia. Combination therapy resulted in overall response rate 24 % with median duration of response 12 months [51].
A humanized anti-CD20 mAb ocrelizumab differs from rituximab at the complementarity-determining regions (CDRs), and shows lower complement dependent cytotoxicity (CDC) but superior ADCC capacity as well as enhanced binding to the low-affinity variants of FcγRIIIa [52]. Ocaratuzumab (LY2469298, AME-133v) is a humanized IgG1 anti-CD20 mAb engineered to have increased affinity to CD20 and enhanced effector function in ADCC assays than rituximab. This regimen in pretreated FL patients who were low-affinity FcyRIIIa allele carriers, was generally well tolerated and common related adverse events included chills and fatigue.
A partial or complete response (CR or PR) was observed in 22 % with a median progression-free survival (PFS) of 25.4 weeks. Ocaratuzumab showed a greater binding affinity for CD20 and enhanced ability in mediating ADCC than rituximab [35].
A novel humanized anti-CD20 antibody BM-ca that binds to distinct epitope 156–166 is most effective in ADCC and anti-cell proliferation activities compared with rituximab and ofatumumab [53]. To exploit this, data from phase I/II study of 12 patients with NHL suggested the drug had promising preliminary anti-lymphoma effects, with 2 CR and 2 PR (33 %), and well tolerated [34].
Development of bispecific antibodies
Bispecific antibodies (bsAb) offer a promising approach of therapeutics over the next decade or so. These antibody formats consisting two different binding arms (e.g., chemical cross-linking or quadromas technique) that are able to redirect immune cells to the tumor site and induce specific tumor cell killing. Treatment of immunodeficient mice bearing human NHL xenografts with CD20-CD22 bsAb (Bs20x22) led to improvements in overall survival (OS) and the reduction of tumor volume when compared to administration of each parent mAb alone or combination rituximab and HB22.7 [54]. Of particular note, targeting both CD20 and tyrosine kinase receptor FMS-related tyrosine kinase 3 (FLT3) (CD20-Flex) by using bsAb was linked to infiltration of dendritic cells (DCs) and cytotoxic T cells (CTLs) into tumor tissues, which could provide long-term protection against recurrence of a tumor [55]. CD47 × CD20 bsAb, that has reduced affinity for CD47 relative to the parental antibody, provided selective phagocytosis of NHL cells and recapitulate the synergy of combination therapy [56].
BsAbs normally activate only a single class of accessory cell and do not induce a long-lasting protective immunity that is thought to lie in the lack of appropriate intact Fc part. Therefore efforts to further increase therapeutic efficacy of these antibody formats resulted in the development of trifunctional bispecific antibodies (trAb) by using the Dock and Lock (DNL) technology to create antibodies with higher valency [57].
BsAbs are also being used to simultaneously retarget of various effector cells such as T cells, NK cell, and macrophage. In agreement, Bi20 (FBTA05), a CD3 x CD20 trAb, has been generated using the quadroma method that preferentially binds to human activating FcγR+ accessory immune cells via its Fc region. Bi20 was shown to be very efficient in the in vitro eliminatation of Raji and NHL cell lines at antibody concentrations as low as 0.5 ng/ml without the need for any additional costimulation. Furthermore this agent mediates effective cytotoxicity against patient derived CLL cells despite increased apoptosis resistance and low CD20 expression levels [58].
In efforts to bring NK cells and tumor cells in proximity, [(CD20)2xCD16] has been constructed to target both CD20 and FcγRIIIA (CD16). The designed novel tribody showed improved clearance of neoplastic B cells compared to rituximab both in vitro and in vivo, suggesting that this antibody formatmay represent promising candidates for clinical applications [59].
Engineering antibody heavy chains for heterodimerization seem likely to emerge as a successful strategy to overcome bsIgG heavy chain-pairing problem.
The homodimerization of two heavy chains in an IgG is mediated by introducing different mutations into two CH3 domains (e.g., knobs-into-holes strategy), resulting in asymmetric antibodies. Anti-CD20/CD3 T cell-dependent bsAb (CD20-TDB) comprises a full-length, humanized immunoglobulin G1 molecule with near-native antibody architecture constructed using “knobs-into-holes” technology. CD20-TDB antibody showed superior anti-lymphoma/leukemia activity both in vitro and in vivo even in the presence of high concentrations of rituximab [60]. Double-variable domain (DVD)-Igs belong to symmetric bsIgG and IgG-like molecules that have reached clinical development. DVD-Ig molecules have been generated from two parent antibodies by appending VL and VH domains of an IgG with the second via short peptide linkers [61]. The recent creation of an anti-CD20/human leukocyte antigen (HLA)-DR DVD-Ig is expected to have the added benefit of inducing ADCC and CDC in vitro, demonstrating the potential of a tetravalent IgG-like molecule as an alternate means to target B-cell lymphoma therapeutically [62].
To improve cross-linking, two novel bispecific hexavalent antibodies (bsHexAbs) (HexAbs: IgG (Fab)4) namely 20-(74)-(74), 74-(20)-(20) derived from veltuzumab/milatuzumab were developed through the DNL method. Besides the potent cytotoxicity of both 20-(74)-(74) and 74-(20)-(20) antibodies against MCL cell lines as well as in primary tumor cells from patients with MCL or CLL, these bsHexAbs could also extended the survival of nude mice bearing MCL xenografts [63].
Anti-CD20 antibody conjugates
Radioimmunoconjugates targeting CD20 antigen, RICs
A different way to improve antitumor efficacy of mAbs is to attach them to radioactive and cytotoxic compounds, known asimmunoconjugates.
Lymphoma cells are inherently radiosensitive; therefore, the anti-CD20 radiolabeled antibodies as the first group of immunoconjugates may provide a significant and durable response advantage in comparison to naked (unlabeled) anti-CD20 mAbs [64].
The approved RICs, anti-CD20 mAbs 90Y-ibritumomab tiuexetan (Zevalin) and 131I-tositumomab (Bexxar) have been used as single reagent or in combination with high-dose chemotherapy (as conditioning regimen) for the treatment of patients with relapsed/refractory low-grade B-cell NHL or FL [65]. The development of RIC with alpha-emitting radionuclides is attractive treatment option for B-cell NHL because of the high linear energy transfer (LET) and shorter path length in human tissue than the more commonly used beta-radiation, allowing higher specific tumor cell kill and lower toxicity to healthy tissues. The in vitro benefit of Bi-labeled rituximab has been further underlined by a recent study using stable99mTc carbonyl diethylene triamine penta acetic acid (DTPA)–rituximab conjugate that showed specificity and receptor affinity for CD20 antigen on Raji cells, indicating its potential for further evaluation as a purely NHL diagnostic agent [66]. Advanced in the treatment era is pretargeted radioimmunotherapy (PT-RAIT), developed by the DNL technology against CD20 for the treatment of NHL. The explanation could lie in the fact, as Altieri performed in xenografted nude mice that compared TF4, a humanized tri-Fab bsAb with two Fabs binding CD20 and one Fab binding histamine-succinyl-glycine (HSG), a pretargeting agent of an 111In-HSG-peptide with the directly conjugated 90Y-anti-CD20 IgG. The DNL tri-Fab recombinant construct demonstrated a significant improved antitumor response with a >40 % cure rate over that of the 90Y-anti-CD20 IgG directly radiolabeled anti-CD20 IgG [67].
Anti-CD20-based cytotoxic drugs or toxins, ADCs
Antibodies have also been coupled to highly potent cytotoxic drugs or toxins (ADCs) to provide a foothold for drug delivery to corresponding antigen-positive cancer cells so as to increase their cytotoxicity and decrease systemic drug exposure. One such agent is rituximab- valine-citrulline antimitotic monomethyl auristatin E (vcMMAE), which appended the anti-CD20 properties of rituximab in lymphoma xenograft models compared with mAb alone [68]. Similar results were also obtained in CD20-positive mice bearing Ramos or Daudi tumor xenografts after exposure to ofatumumab-vcMMAE [69]. In addition, studies of rituximab-type I ribosome-inactivating protein (RIP) saporin when combined with chemotherapy was efficacious against Raji cell lines [70].
Anti-CD20 antibody-cytokine fusion protein, ICs
Another important research field is development of antibodies by attaching cytokines to their Fc terminus, called immunocytokines (ICs) to mediate greater NK-cell ADCC through a higher affinity for CD16/FcγRIIIa. With efforts to increase the stimulation activity of soluble IL15, a fusion protein linking IL15Rα chain sushi domain and human IL15 (RLI) has been generated. In agreement, a recent study using RLI-RTX (rituximab) in mouse models of lymphoma, the long-term survival versus either agent alone or in combination was found to correlate with improved clinical efficacy of RTX [71]. In addition to these treatment modalities, Rossi et al. developed tetrameric IC comprising 4 Interferon-α2β (IFN-α2βα) groups site-specifically attached to veltuzumab via the DNL method. The potent therapeutic efficacy of mAb-IFN-α ICs against human lymphoma xenografts models compared favorably in activity with equidoses of the parent mAb, the cytokine or an irrelevant IgG-IFN conjugate [72]. In vitro experience with a bispecific IC 20-C2-2β consisting of two copies of IFN α2β and a stabilized F (ab)2 of HLA-DR-targeting humanized antibody hL243 has shown that this conjugate could effectively deplete a wide variety of malignant B cells [73]. A recombinant anti-CD20-human interleukin-2 (hIL-2) IC (2B8-Fc-hIL2) based on the therapeutic rituximab antibody scaffold expressed in Nicotiana benthamiana could be effective in eliciting ADCC of lymphoma cells [74]
CD20 targeted therapy using nanotechnology
As an alternative to antibodies, multiple studies have focused on providing selective delivery of therapeutic agents harbored by nanoparticle (NP) platforms. There is an interesting body of work supported an in vitro significant decrease of BCL-2 protein expression level when the anti-CD20 antibody conjugated to gold nanoparticles (AuNP) through the spontaneous reaction of the cysteine thiol groups [75].
A recent study using anti-CD20 coated gold NP and irradiating by a few resonant femtosecond pulses in vitro was able to link high level of intracellular reactive oxygen species (ROS) to apoptosis within several hours [76].
Guided by Voltan et al. work to develop NP for treatment of SCID mice with a JVM-2 (p53wild-type B-leukemic cells) xenografts, they designed three construction including Nutlin-3 as an anti-cancer agent in a variety of p53wild-type tumors encapsulated into poly lactide-co-glycolide NPs (NP-Nut), rituximab-engineered NPs (NP-Rt-Nut) and NPs engineered with rituximab alone (NP-Rt). Of these, however, NP-Rt-Nut prolonged the survival rates of mice more efficiently than NP-Nut and/or NP-Rt [77]. The concept that antibody-conjugated immunoliposomes might be a very promising delivery system has also been reported in mice bearing human B lymphoma in which CD20-targeted pegylated liposomal vincristine (VCR) significantly prolonged survival rates compared with free VCR CD20-targeted liposomal doxorubicin (DXR). Additionally, 70 % remission rates through anti-CD19 and anti-CD20-targeted liposomal VCR have been described in this study [78].
Owing to its lower cardiotoxic potential, nonpegylated liposomal doxorubicin (NPLD) is given in combination with rituximab, cyclophosphamide, vincristine, and, prednisone (R-COMP) as a first-line treatment for splenic marginal zone lymphoma (SMZL) leading to an ORR of 84 % [79, 80]. It is now well documented that rituximab-conjugated liposomal G3139 (an antisense ODN against Bcl-2) NPs could enhance in vitro sensitivity of CLL cells to fludarabine and promote significant in vivo antitumor activity [81]. Strikingly, the basic research program elaborated N-(2-hydroxypropyl) methacrylamide (HPMA) copolymer conjugates by using modern controlled radical polymerisation technique, reversible addition fragmentation chain transfer (RAFT) appears to be effective against B-cell neoplasia. Support for these beneficial effects comes from a recent study suggesting that multivalent HPMA copolymer conjugates attached to multiple Fab’ fragments of the anti-CD20 mAb (1F5) exert significant avidity and apoptotic activity during longer exposure times compared to unconjugated whole mAb [82].
Cellular immunotherapy for B-cell malignancies
Adoptive transfer of endogenous T cells genetically modified to express a chimeric antigen receptor (CAR) targeting CD20 antigen is an expanding area of interest within the field of immunotherapy [83–85] (Table 3). Early generation CAR bearing a CD20-targeted single-chain variable fragment (scFv) and one signaling domain CD3ζ (scFvFczeta) demonstrated antigen-specific IL-2 production as well as cytolytic activity against human lymphoma in vitro [86]. These results were confirmed in studies with engineered human cytotoxic T lymphocytes (CTL) containing first generation CAR-CD20 which showed antigen-dependent interferon gama (IFN-γ) secretion upon co-culture with CD20+ lymphoma stimulator cells. Such CTL exhibit specific cytotoxicity against actual tumor cells isolated from patients with a variety of B lymphoid malignancies [87]. Wang et al., constructed a second-generation CARs endowed with costimulatory endodomains CD28 and a third-generation CAR containing a co-stimulatory domain derived from CD28, CD137 (4-1BB) in order to redirect them to tumor cells. These αCD19-BB-ζ CARs were capable of promoting anti-lymphoma activity in vitro compared with T cells expressing first generation CARs [88]. Specifically, a third-generation 1F5 (anti-CD20)-CAR containing an inducible iC9 and a truncated CD19 (Δ19) selectable marker has likewise demonstrated elimination of more than 90 % of the iC9-CD20 CAR- Δ19 T cells upon activation of iC9 both in vitro and in vivo [89]. As an alternative to T cells, NK cells have also been found to be suitable effectors for expressing CARs directed against CD20 in cell-based immunotherapy. Accordingly, NK cells transduced with a chimeric receptor specific for CD20 share the capacity to mediate increased cytotoxic activity against primary lymphoma and leukemia cell lines as compared with the control [90]. Similarly, the anti-CD20 CAR-expressing NK-92 cells were found to be superior in controlling local tumor growth of some neoplastic B-lymphoid xenografts in immunocompromised mice [91]. The preclinical relevance of expanded peripheral blood NK cells (exPBNK) modified with anti-CD20 CAR following mRNA nucleofection has been further underlined by the demonstration that CAR+ exPBNK can significantly extended survival time (P < 0.001) and reduce tumor size in human burkitt lymphoma xenografted NOD/SCID/γ-chain−/− (NSG) mice [92]. Stem cell-based therapies are emerging as a promising strategy to tackle cancer. Multiple stem cell types have been shown to exhibit inherent tropism towards tumors. Moreover, when engineered to express therapeutic agents, these pathotropic delivery vehicles can effectively target sites of malignancy. This perspective considers the current status of stem cell-based treatments for cancer and provides a rationale for translating the most promising preclinical studies into the clinic. Mesenchymal stem cells (MSCs) transduced with anticancer peptides are currently being explored for site-specific drug delivery to inhibit tumor growth and progression. However, there are efforts to overcome tumor-supportive properties and off-target effects of MSCs, in order to make them as an attractive candidate for cell-based therapy [93].
Human umbilical cord derived MSCs (HUMSCs) expressing scFvCD20-soluble tumor necrosis factor-related apoptosis-inducing ligand (sTRAIL) fusion protein have shown potent preclinical efficacy in mice beating lymphoma xenograft [94].
Targeting TNFSFR and CD20 antigen with fusion protein
The activation of tumor necrosis factor superfamily receptors (TNFSFR) by their respective ligands or agonistic mAbs have been proposed as being a potential target for cancer immunotherapy to induce tumor cell apoptosis via extrinsic pathway. Interestingly, genetically fusing a rituximab-derived antibody fragment to soluble FasL (scFv:sFasL) has been shown to trigger apoptotic elimination of malignant B-cells upon CD20 cross-linking [95]. To explore targeted delivery of soluble trimeric CD40L (sCD40L) to the B cell leukemia marker CD20 as a therapeutic option, scFvCD20:CD40L was generated. The scFvCD20:CD40L-based fusion protein augmented effective paracrine maturation of DCs and also triggered a significant decrease of cell viability in certain leukemic B cell lines as a result of CD20-induced apoptosis [96].
Antibody-based combination therapy
Immunotherapy followed by stem cell transplantation
Tumor-specific mAbs when used in vivo in conjunction with either autologous or allogeneic hematopoietic stem cell transplantation (HSCT) represents a completely different approach for the treatment of chemosensitive relapsed patients. Results from several studies have reported that the addition of rituximab to second-line chemotherapy concurrent with autologous hematopoietic stem cell transplantation (ASCT) improves PFS than patients do not receive rituximab in their first-line treatment. Accordingly, Magni et al., presented data from 15 patients with relapsed indolent lymphoma or MCL who were treated with a combination of rituximab and high-dose chemotherapy followed by ASCT. The efficacy of marrow treatment was evaluated by PCR for the presence of t (14;18) translocation before and after the purge. The results indicated that all patients transplanted in this study had become negative for the translocation and achieved at least a complete remission from induction therapy. Although a plethora of study associated with encouraging results of maintenance rituximab after ASCT have been described, the findings of Gisselbrecht,s group revealed no significant survival advantage in refractory DLBCL patients [97]. Furthermore, recent studies established a link between the incorporation of radioimmunotherapy in reduced-intensity conditioning (RIC) allo-HSCT regimens and favorable outcome. The main reason for this successful clinical translation lies in Dana Farber Cancer Institute study which published the results of a prospective trial of patients with relapsed, refractory or transformed FL who received yttrium-90 (90Y)-ibritumomab tiuxetan followed by fludarabine and low-dose busulfan RIC allogeneic HSCT. The outcomes of 41 patients with FL were considerably resulted in the 2-year OS, PFS and nonrelapse mortality (NRM) were 83, 74, and 18 %, respectively. Based on these results the incorporation of radioimmunotherapy (RIT) in RIC allo-HSCT regimens is feasible because of the excellent OS and PFS and acceptable rates of graft-versus-host disease (GVHD) and relapse compared with regimens that do not include RIT [98].
Cytokines combined with rituximab
Recent data support the concept that cytokines are the critical cause of leukemic blast cell differentiation into professional antigen-presenting cells initiating antitumor immune responses that make it a better target for immunotherapy. These investigations raise the possibility that GM-CSF administered with R-CHOP (rituximab-cyclophosphamide, vincristine, doxorubicin and prednisolone) can modestly improve survival of previously untreated DLBCL in the elderly [99]. In this regard, phase II study of rituximab plus GM-CSF in patients with FL has already been completed, but no results were reported [NCT01939730]. It was further observed that the L19 antibody (specific for alternatively-spliced extra-domain B (EDB) of fibronectin)-mediated delivery of IL-2 to tumor microenvironment isolated from phage display libraries was able to completely eradicate B-cell lymphoma xenografts when combined with rituximab [100]. Besides these cytokines, a fusion protein consisting of IL15 mutant and IL-15Rα/Fc complex could also improve rituximab-mediated ADCC against B cell lymphoma both in vitro and in vivo [101]. Therapeutic effects of ALT-803, an IL-15 Superagonist, in combination with rituximab is currently under clinical trial for relapsed/refractory iNHL [NCT02384954]. Moreover, the efficacy of rituximab plus recombinant IL-21 was evaluated in a phase I trial, reporting durable complete remissions in the subset of patients with relapsed and refractory low grade B-cell lymphoproliferative disorders [102].
Dendritic cell vaccine-based immunotherapy
Immunotherapeutic strategies have attempted to monopolize on the efficient antigen-presenting capabilities of DCs to deliver ligands for activation of specific effector T cells as a means of therapeutic vaccination in patients with advanced cancers. Recent data have shown that the combination of rituximab and DC vaccine induces the generation of T-cell responses, leading to long-lasting antitumor immune protection in syngeneic mouse lymphoma models [103]. Manzur et al., further highlighted that an enhanced therapeutic effect of B cell-depleting anti-CD20 mAbs in lymphoma-bearing mice when combined with DC vaccination might be correlated with promotion of antigen cross-presentation, induction of a protective T cell response, and increased injected DCs activation through reducing of tumor-derived IL-10 [104].
Exosome removal as a therapeutic adjuvant for lymphoma/leukemia immunotherapy
Extensive analyses by various techniques have suggested that exosome formation derived from tumor cells plays important roles in regulating all facets of cancer development and spread [105]. Activation of BCR triggers the release of CD20-bearing tumor exosomes where they bind to anti-CD20, which protect target cells from antibody attack [106]. In addition to interfering with the activity of immunotherapeutic agents, exosomal survivin, the inhibitor of apoptosis (IAP) protein family, participate in skewing T helper populations (Th1/Th2) and decreasing their cytotoxic function [107].
Interestingly, lymphoma-derived exosomes were found to be modulated by lysosome-related, organelle associated ATP-binding cassette transporter A3 (ABCA3) both in vitro and in vivo. Consistent with this observations, the pharmacologic silencing of ABCA3 using the cyclooxygenase type-2 inhibitor indomethacin abrogated exosome biogenesis, thereby reducing exosome-mediated shielding of target cells and increasing the anti-tumor efficacy of rituximab [108].
Therapeutic agents targeting cellular response to stress and combinations
It has become increasingly evident that transformed cells allow acquiring a resistance to increased cellular stress conditions in order to ensure cell survival and many homeostatic functions. Depletion of significant survival proteins such as Akt and cyclin D1 and thereby producing antitumour effects in a wide range of lymphoma cell lines by pharmacologic disruption of homeostatic molecules such as a ubiquitously expressed chaperone, heat shock protein 90 (HSP90), has gained more interest in recent years [109, 110]. It is commonly believed that the ubiquitin-proteasome pathway is the major process for the non-lysosomal intracellular proteins degradation also used to maintain homeostasis.
With regard to treatment, proteasome suppressant have been shown to be critical for the inhibition of NF-κB activation which may also prevent angiogenesis and metastasis in vivo and further increase the sensitivity of cancer cells to apoptosis [111].
The induction of apoptosis by proteasome inhibitor, bortezomib occurs following elevated apoptotic protease-activating factor-1 (Apaf-1) expression in human leukemia cells [112].
Supporting a potent role for proteasome inhibitors in cancer cells, the results from the phase II study in elderly MCL patients clearly confirmed the benefit of adding bortezomib to rituximab, doxorubicin, dexamethasone, and chlorambucil (RiPAD + C) as a first-line therapy [113]. In another phase I/II study bortezomib plus R-CHOP appears to produce clinical advantage for non- germinal center B cell (GCB) DLBCL subtype by enhanced activity of dose-adjusted chemotherapy [114].
The mechanism by which leukemia cells were protected against apoptosis induced by proteasome inhibitors seems to be related to BCL-2 overexpression [115]. To overcome resistance, the potential clinical administration of bortezomib, rituximab, and obatoclax, a novel pan-BCL-2 inhibitor, has been shown to be effective in killing of both rituximab-sensitive (RSCL) and rituximab-resistant B-cell lymphoma cell lines (RRCL) [116].
In CLL cells, Hsp90 broadly functions can be reversed with 17-allylamino-17-demethoxygeldanamycin (17-DMAG) where it show synergistic cytotoxicity with rituximab [117]. SNX-2112, an oral Hsp-90 Inhibitor, was found to exert augmented antitumor activity when added to rituximab and bortezomib in rituximab-resistant NHL cells lines, which make this way into the clinic trial [118].
Triggering TRAIL/TRAILR along with rituximab
Within the TNF superfamily, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has gained prominence as possible therapeutic modulators owing to its tumoricidal activity in animal models and in a variety of tumor types, with no or minimal toxicity towards normal cells [119]. Based on this promising activity profile, the administration of mapatumumab, a fully human agonistic mAb directed to TRAIL receptor 1 (TRAIL-R1) and rituximab has shown enhanced antilymphoma efficacy in preclinical models [120]. Dulanermin, a soluble recombinant human apoptosis ligand 2 (Apo2L) or TRAIL, was found to be safe and well tolerated when combined with rituximab in NHL patients [121].
However, an open-label phase 1b/2 randomized study revealed no added benefit of this combined treatment in relapsed patients with iNHL [122].
Cell cycle regulators as targets for therapy and combinations
Dysregulation of cell cycle process is one of the hallmarks of cancer, making it an attractive targets for pharmacological inhibition. Given the potential benefits of cyclin dependent kinase (CDK) inhibitors, a study of cyclophosphamide plus alvocidib, a CDK inhibitor, and rituximab (CAR) is enrolled for treatment of nine high-risk CLL patients. Combined therapy with CAR was well tolerated, resulted in three CR and four PR [123]. In a phase I trial, the combination of dinaciclib, a novel selective inhibitor of cyclin-dependent kinase (CDK1, 2, 5, and 9) with rituximab in relapsed/refractory CLL subjects, was well tolerated and only one patient achieved a CR [124]. Another Phase I study involved dinaciclib plus rituximab for CLL and SLL patient [NCT01650727]. A high Skp2, a p27 (kip1) ubiquitin ligase, expression was found to be correlated with poor OS and PFS in DLBCL patients who received R-CHOP regimen [125].
Overexpression of Aurora A can override the spindle assembly checkpoint activity during the mammalian cell cycle and lead to resistance to microtubule-targeted drugs (MTA, e.g., taxanes, vinca alkaloids) treatment. Indeed, several studies have been designed to assess efficacy of Aurora kinase as single agents or combined with other regimens. Xenograft experiment using MCL cell lines has shown synergistic therapeutic benefit by combining MLN8237, an Aurora A inhibitor, with rituximab and MTA (e.g., docetaxel or vincristine) [126]. In B cell malignancies, several clinical trials are currently testing MLN8237 in combination with rituximab and/or other therapeutic agents (e.g., bortezomib or vincristine) [NCT01812005, NCT01397825, NCT00651664].
Treatment of DLBCL cell lines with mammalian target of rapamycin inhibitor everolimus plus rituximab induces G0/G1 cell cycle arrest and decrease the overexpression of p-AKT, demonstrating a rationale for potential synergistic effect [127].
Of greit interest, combined administration of ON 013105, a novel benzylstyryl sulfone kinase inhibitor, and rituximab was found to inhibit MCL proliferation by modulating myeloid cell leukemia 1 (Mcl-1) expression, an essential protein for cell growth and survival [128].
For MCL cohorts (based on CD20 and cyclin D1-positive cells) the addition of BTK inhibitor ibrutinib to rituximab is active and well tolerated [129]. Combination therapy with idelalisib as a first-in-class, delta isoform specific, PI3-kinase inhibitor plus rituximab had durable disease control in treatment-naïve older patients with CLL. The documented ORR was 100 % in patients with deletion (17p)/TP53 mutations and 97 % in those with unmutated IGHV and a median PFS of 36 months [130]. Duvelisib, a novel PI3K-δ,γ inhibitor, has also been incorporated into rituximab or obinutuzumab in subjects with previously untreated CD20+ FL [NCT02391545].
Immunomodulatory drugs-rituximab combinations
Using DLBCL cell lines, lenalidomide treatment was found to significantly inhibit proliferation of activated B cell–like (ABC)-DLBCL in vitro, by blocking interferon regulatory factor 4 (IRF4) expression and the BCR-NF-κB signaling pathway in a cullin 4-containing ubiquitin ligase complex (CRL4)-dependent manner [131].
Fecteau et al., showed that lenalidomide can directly inhibit proliferation of CLL cells in a cereblon- and p21-dependent, but p53-independent manner [132].
These studies provided the basis for the use of lenalidomide combined with rituximab in NHL, demonstrating its antitumor effects via the recruitment of NK cells, increased ADCC, improved NK cell–mediated synapse formation, and CD20 capping [133–137]. Fowler et al., presented data using lenalidomide combined with rituximab for untreated indolent lymphoma. Treatment was well tolerated, with an ORR of 98 %, and PFS of 89 %. [138]. Immunologic effects of lenalidomide through reduction of Tregs number and function may potentiate the rituximab-mediated ADCC action in patients with FL who are FcγRIIIa-158 phenylalanine carriers, resulted in ORR of 46 % with a 2-year PFS of 44 % [139].
In a phase II study lenalidomide-rituximab combination showed an ORR of 84 % with 53 % CR in patients with MCL [140]. The feasibility of combining lenalidomide concurrently with rituximab and bortezomib has been demonstrated in phase I/II trials of MCL patients. The three-drug therapy resulted in a 82 % ORR with 32 % of patients having a CR and 75 % ORR for patients who did not received prior treatment The authors suggest that, given the incidence of neuropathy, subcutaneous or less frequent intravenous (IV) dosing of bortezomib should be considered rather than the twice-weekly IV bortezomib used in this study [141].
A novel immunomodulatory drugs (IMiDs) CC-122, the pleiotropic pathway modifier, specifically targets CRL4, and promotes degradation of Aiolos and Ikaros, resulting in a mimicry of IFN signaling and apoptosis in DLBCL [142]. Consequently, ongoing study is assessing CC-122 combined with rituximab and two other IMiDs, CC-223, CC-292, in subjects with DLBCL [NCT02031419].
Histone deacetylase inhibitor-containing drugs
Epigenetic code modifications including altered DNA methylation and histone acetylation have been proposed as promising therapeutic targets in B cell malignancies. Interestingly, recent work has shown that histone deacetylase inhibition (HDACi) is associated with increased CD20 expression level in DLBCL patients in vivo, suggesting that pre-treatment with valproate or other HDACis before anti-CD20 therapy could be beneficial in CD20-low B-cell lymphomas [59, 143]. In Phase I/II trial setting, the combination of vorinostat (SAHA) histone de-acetylases (HDACi), cladribine, and rituximab (SCR) for newly diagnosed MCL produced considerable response rate and clinical benefit, perhaps correlating with ADCC activity, CD20 mRNA expression level, and cyclin D1-A polymorphism [144, 145]. Another trial of SCR established in patients with relapsed B-cell malignancies. This compound revealed a trend towards decreased expression of multiple tumor suppressor genes including DUSP1, DUSP 2, and p53 as well as changes in CD20 mRNA levels, and was well tolerated especially in patients with previously untreated MCL [146]. The vorinostat-rituximab combination was found to be considerably safe and active in iNHL with ORR of 46 %. Although the downregulation of cytokines after treatment with vorinostat and rituximab demonstrated no increase in formal response, it is possible that analysis of various cytokines secretion at different time points would enhance the correlation with response outcomes [147]. Another HDACi, panobinostat, is being evaluated in combination with rituximab for relapsed/refractory DLBCL [NCT01238692].
Other new combined reagents
A broad range of other novel drugs with various mechanisms of action are still under development or in early stage of clinical studies. Emerging in vitro and in vivo experimental settings suggested the involvement of fenretinide (4HPR), synthetic analog of retinoic acid (RA), and rituximab not only in apoptosis induction of malignant human B cells but also in elimination of established tumors and prolongation of survival times in mice bearing B lymphoma xenografts, favoring combination arm [148]. Tablostat mesylate exerts inhibitory effects on dipeptidyl peptidase such as fibroblast activation protein (FAP) and dipeptidyl peptidase IV (DPP-IV) and stimulates antineoplastic and haematopoesis activities. In xenograft models of human CD20 + B-cell lymphoma talabostat enhanced the cytotoxic efficacy of rituximab [149]. Accordingly, phase II trial of tablostat plus rituximab in CLL patient who relapse after, or do not respond to fludarabine regimen has completed; although no results have been reported [NCT00086203].
Lysophosphatidic acid acyltransferase β (LPAAT-β) isoform is a transmembrane enzyme that catalyses the biosynthesis of phosphatidic acid and stimulates a specific G protein–coupled receptor present in numerous cell types, thereby triggering a multitude of biologic responses. Enzymatic inhibition of LPAAT-β in combination with rituximab has been linked to antitumor activity in NHL cell lines and induction of complete response in human lymphoma xenografts [150]. Importantly, a recent study have provided interesting clues illustrating farnesyltransferase inhibitor (FTIs) L-744,832, which reportedly inhibits prosurvival signaling, has capacity to increase anti-tumor activity of rituximab and CD20 expression in the majority of primary NHL or CLL cells [151].
Conclusions
Although clinical outcome for leukemia and lymphoma with CD20 mAbs has a considerable improvement over the last decade, the development of resistance in certain patients is still a major problem. Therefore, new generations of anti-CD20 mAbs, antibody engineering approaches and antibody combination therapies with superior inhibitory activity provide extensive opportunities for treatment of B cell neoplasia.
References
Loken MR, Shah VO, Dattilio KL, Civin CI (1987) Flow cytometric analysis of human bone marrow. II. Normal B lymphocyte development. Blood 70:1316–1324
Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, Beaumont T, Tedder TF, van Noesel CJ, Eldering E, van Lier RA (2010) CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest 120:214–222
Tedder TF, Engel P (1994) CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today 15:450–454
Winiarska M, Bil J, Nowis D, Golab J (2010) Proteolytic pathways involved in modulation of CD20 levels. Autophagy 6:810–812
van Meerten T, Hagenbeek A (2009) CD20-targeted therapy: a breakthrough in the treatment of non-Hodgkin’s lymphoma. Neth J Med 67:251–259
Farajnia S, Ahmadzadeh V, Tanomand A, Veisi K, Khosroshahi SA, Rahbarnia L (2014) Development trends for generation of single-chain antibody fragments. Immunopharmacol Immunotoxicol 36:297–308
Cragg MS, Walshe CA, Ivanov AO, Glennie MJ (2005) The biology of CD20 and its potential as a target for mAb therapy. Curr Dir Autoimmun 8:140–174
Polyak MJ, Tailor SH, Deans JP (1998) Identification of a cytoplasmic region of CD20 required for its redistribution to a detergent-insoluble membrane compartment. J Immunol 161:3242–3248
Binder M, Otto F, Mertelsmann R, Veelken H, Trepel M (2006) The epitope recognized by rituximab. Blood 108:1975–1978
Tedder TF, Schlossman SF (1988) Phosphorylation of the B1 (CD20) molecule by normal and malignant human B lymphocytes. J Biol Chem 263:10009–10015
Ivaldi C, Martin BR, Kieffer-Jaquinod S, Chapel A, Levade T, Garin J, Journet A (2012) Proteomic analysis of S-acylated proteins in human B cells reveals palmitoylation of the immune regulators CD20 and CD23. PLoS One 7:e37187
Giles FJ, Vose JM, Do KA, Johnson MM, Manshouri T, Bociek G, Bierman PJ, O’Brien SM, Keating MJ, Kantarjian HM, Armitage JO, Albitar M (2003) Circulating CD20 and CD52 in patients with non-Hodgkin’s lymphoma or Hodgkin’s disease. Br J Haematol 123:850–857
Deans JP, Li H, Polyak MJ (2002) CD20-mediated apoptosis: signalling through lipid rafts. Immunology 107:176–182
Li H, Ayer LM, Lytton J, Deans JP (2003) Store-operated cation entry mediated by CD20 in membrane rafts. J Biol Chem 278:42427–42434
Vugmeyster Y, Howell K, Bakshl A, Flores C, Canova-Davis E (2003) Effect of anti-CD20 monoclonal antibody, Rituxan, on cynomolgus monkey and human B cells in a whole blood matrix. Cytom A 52:101–109
Al-Zoobi L, Salti S, Colavecchio A, Jundi M, Nadiri A, Hassan GS, El-Gabalawy H, Mourad W (2014) Enhancement of Rituximab-induced cell death by the physical association of CD20 with CD40 molecules on the cell surface. Int Immunol 26:451–465
Rohrbach P, Broders O, Toleikis L, Dubel S (2003) Therapeutic antibodies and antibody fusion proteins. Biotechnol Genet Eng Rev 20:137–163
Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Wang Y, Chang CH (2008) Novel designs of multivalent anti-CD20 humanized antibodies as improved lymphoma therapeutics. Cancer Res 68:8384–8392
Henry C, Deschamps M, Rohrlich PS, Pallandre JR, Remy-Martin JP, Callanan M, Traverse-Glehen A, GrandClement C, Garnache-Ottou F, Gressin R, Deconinck E, Salles G, Robinet E, Tiberghien P, Borg C, Ferrand C (2010) Identification of an alternative CD20 transcript variant in B-cell malignancies coding for a novel protein associated to rituximab resistance. Blood 115:2420–2429
Beers SA, French RR, Chan HT, Lim SH, Jarrett TC, Vidal RM, Wijayaweera SS, Dixon SV, Kim H, Cox KL, Kerr JP, Johnston DA, Johnson PW, Verbeek JS, Glennie MJ, Cragg MS (2010) Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115:5191–5201
Reslan L, Dalle S, Dumontet C (2000) Understanding and circumventing resistance to anticancer monoclonal antibodies. MAbs 1:222–229
Racila E, Link BK, Weng WK, Witzig TE, Ansell S, Maurer MJ, Huang J, Dahle C, Halwani A, Levy R, Weiner GJ (2008) A polymorphism in the complement component C1qA correlates with prolonged response following rituximab therapy of follicular lymphoma. Clin Cancer Res 14:6697–6703
Treon SP, Hansen M, Branagan AR, Verselis S, Emmanouilides C, Kimby E, Frankel SR, Touroutoglou N, Turnbull B, Anderson KC, Maloney DG, Fox EA (2005) Polymorphisms in FcgammaRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenstrom’s macroglobulinemia. J Clin Oncol 23:474–481
Golay J, Zaffaroni L, Vaccari T, Lazzari M, Borleri GM, Bernasconi S, Tedesco F, Rambaldi A, Introna M (2000) Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood 95:3900–3908
Boross P, Leusen JH (2012) Mechanisms of action of CD20 antibodies. Am J Cancer Res 2:676–690
Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, van Meerten T, Ebeling S, Vink T, Slootstra JW, Parren PW, Glennie MJ, van de Winkel JG (2006) The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol 177:362–371
Nishida M, Uematsu N, Kobayashi H, Matsunaga Y, Ishida S, Takata M, Niwa O, Padlan EA, Newman R (2011) BM-ca is a newly defined type I/II anti-CD20 monoclonal antibody with unique biological properties. Int J Oncol 38:335–344
Alduaij W, Ivanov A, Honeychurch J, Cheadle EJ, Potluri S, Lim SH, Shimada K, Chan CH, Tutt A, Beers SA, Glennie MJ, Cragg MS, Illidge TM (2011) Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 117:4519–4529
Chan HT, Hughes D, French RR, Tutt AL, Walshe CA, Teeling JL, Glennie MJ, Cragg MS (2003) CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Res 63:5480–5489
Cragg MS, Morgan SM, Chan HT, Morgan BP, Filatov AV, Johnson PW, French RR, Glennie MJ (2003) Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 101:1045–1052
Cragg MS, Glennie MJ (2004) Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood 103:2738–2743
Mossner E, Brunker P, Moser S, Puntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E, Ferrara C, Sondermann P, Jager C, Strein P, Fertig G, Friess T, Schull C, Bauer S, Dal PJ, Del NC, Dabbagh K, Dyer MJ, Poppema S, Klein C, Umana P (2010) Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 115:4393–4402
Niederfellner G, Lammens A, Mundigl O, Georges GJ, Schaefer W, Schwaiger M, Franke A, Wiechmann K, Jenewein S, Slootstra JW, Timmerman P, Brannstrom A, Lindstrom F, Mossner E, Umana P, Hopfner KP, Klein C (2011) Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 118:358–367
Kensei Tobinai MO, Dai M, Tatsuya S, Yukio K, Toshiki U, Suguru F, Takashi O, Tomoharu F, Yasuhiko K (2013) Phase I study of a novel humanized anti-CD20 antibody, BM-ca, in patients (pts) with relapsed or refractory indolent B cell non-Hodgkin lymphoma (B-NHL) pretreated with rituximab. J Clin Oncol 31:8551
Forero-Torres A, de Vos S, Pohlman BL, Pashkevich M, Cronier DM, Dang NH, Carpenter SP, Allan BW, Nelson JG, Slapak CA, Smith MR, Link BK, Wooldridge JE, Ganjoo KN (2012) Results of a phase 1 study of AME-133v (LY2469298), an Fc-engineered humanized monoclonal anti-CD20 antibody, in FcgammaRIIIa-genotyped patients with previously treated follicular lymphoma. Clin Cancer Res 18:1395–1403
Ganjoo KN, de Vos S, Pohlman BL, Flinn IW, Forero-Torres A, Enas NH, Cronier DM, Dang NH, Foon KA, Carpenter SP, Slapak CA, Link BK, Smith MR, Mapara MY, Wooldridge JE (2015) Phase 1/2 study of ocaratuzumab, an Fc-engineered humanized anti-CD20 monoclonal antibody, in low-affinity FcgammaRIIIa patients with previously treated follicular lymphoma. Leuk Lymphoma 56:42–48
Salles GA, Morschhauser F, Solal-Celigny P, Thieblemont C, Lamy T, Tilly H, Gyan E, Lei G, Wenger M, Wassner-Fritsch E, Cartron G (2013) Obinutuzumab (GA101) in patients with relapsed/refractory indolent non-Hodgkin lymphoma: results from the phase II GAUGUIN study. J Clin Oncol 31:2920–2926
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner C, Chagorova T, De la Serna J, Dilhuydy M, Opat S, Carolyn J, Owen CJ, Samoylova O, Kreuzer K, Anton W, Langerak AW, Ritgen M, Stilgenbauer S, Döhner HD, Asikanius E, Humphrey K, Michael K, Wenger MK, Hallek M (2013) Head-to-head comparison of obinutuzumab (GA101) plus chlorambucil (Clb) versus rituximab plus Clb in patients with chronic lymphocytic leukemia (CLL) and co-existing medical conditions (comorbidities): final stage two results of the CLL11 trial. Blood 122:6
Morschhauser FA, Cartron G, Thieblemont C, Solal-Celigny P, Haioun C, Bouabdallah R, Feugier P, Bouabdallah K, Asikanius E, Lei G, Wenger M, Wassner-Fritsch E, Salles GA (2013) Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large b-cell lymphoma or mantle-cell lymphoma: results from the phase II GAUGUIN study. J Clin Oncol 31:2912–2919
Morschhauser F, Leonard JP, Fayad L, Coiffier B, Petillon MO, Coleman M, Schuster SJ, Dyer MJ, Horne H, Teoh N, Wegener WA, Goldenberg DM (2009) Humanized anti-CD20 antibody, veltuzumab, in refractory/recurrent non-Hodgkin’s lymphoma: phase I/II results. J Clin Oncol 27:3346–3353
Brown JR, O’Brien S, Kingsley CD, Eradat H, Pagel JM, Lymp J, Hirata J, Kipps TJ (2015) Obinutuzumab plus fludarabine/cyclophosphamide or bendamustine in the initial therapy of CLL patients: the phase 1b GALTON trial. Blood 125:2779–2785
Morschhauser F, Marlton P, Vitolo U, Linden O, Seymour JF, Crump M, Coiffier B, Foa R, Wassner E, Burger HU, Brennan B, Mendila M (2010) Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann Oncol 21:1870–1876
Casulo C, Vose JM, Ho WY, Kahl B, Brunvand M, Goy A, Kasamon Y, Cheson B, Friedberg JW (2014) A phase I study of PRO131921, a novel anti-CD20 monoclonal antibody in patients with relapsed/refractory CD20+ indolent NHL: correlation between clinical responses and AUC pharmacokinetics. Clin Immunol 154:37–46
Jak M, van Bochove GG, Reits EA, Kallemeijn WW, Tromp JM, Umana P, Klein C, van Lier RA, van Oers MH, Eldering E (2011) CD40 stimulation sensitizes CLL cells to lysosomal cell death induction by type II anti-CD20 mAb GA101. Blood 118:5178–5188
Yin C, Lee S, O’Connell T, Ayello J, van de Ven C, Cairo MS (2014) Obinutuzumab (GA101) Significantly Inhibits Cell Proliferation and Induces Programmed Cell Death in Primary Mediastinal B-Cell Lymphoma (PMBL): Obinutuzumab May be a Future Targeted Agent for the Treatment of PMBL. Blood 124:4492
Byrd JC, Flynn JM, Kipps TJ, Boxer M, Kolibaba KS, Carlile DJ, Fingerle-Rowson G, Tyson N, Hirata J, Sharman JP (2016) Randomized phase 2 study of obinutuzumab monotherapy in symptomatic, previously untreated chronic lymphocytic leukemia. Blood 127:79–86
Rafiq S, Butchar JP, Cheney C, Mo X, Trotta R, Caligiuri M, Jarjoura D, Tridandapani S, Muthusamy N, Byrd JC (2013) Comparative assessment of clinically utilized CD20-directed antibodies in chronic lymphocytic leukemia cells reveals divergent NK cell, monocyte, and macrophage properties. J Immunol 190:2702–2711
Brychtova Y, Panovska A, Sebejova L, Stehlikova O, Chovancova J, Malcikova J, Smardova J, Plevova K, Volfova P, Trbusek M, Mraz M, Bakesova D, Trizuljak J, Hadrabova M, Obrtlikova P, Karban J, Smolej L, Oltova A, Jelinkova E, Pospisilova S, Mayer J (2015) Ofatumumab added to dexamethasone in patients with relapsed or refractory chronic lymphocytic leukemia: results from a phase II study. Am J Hematol 90:417–421
Goldenberg DM, Rossi EA, Stein R, Cardillo TM, Czuczman MS, Hernandez-Ilizaliturri FJ, Hansen HJ, Chang CH (2009) Properties and structure-function relationships of veltuzumab (hA20), a humanized anti-CD20 monoclonal antibody. Blood 113:1062–1107
Negrea GO, Elstrom R, Allen SL, Rai KR, Abbasi RM, Farber CM, Teoh N, Horne H, Wegener WA, Goldenberg DM (2011) Subcutaneous injections of low-dose veltuzumab (humanized anti-CD20 antibody) are safe and active in patients with indolent non-Hodgkin’s lymphoma. Haematologica 96:567–573
Christian BA, Poi M, Jones JA, Porcu P, Maddocks K, Flynn JM, Benson DM Jr, Phelps MA, Wei L, Byrd JC, Wegener WA, Goldenberg DM, Baiocchi RA, Blum KA (2015) The combination of milatuzumab, a humanized anti-CD74 antibody, and veltuzumab, a humanized anti-CD20 antibody, demonstrates activity in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Br J Haematol 169:701–710
Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H (2002) Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99:754–758
Kobayashi H, Matsunaga Y, Uchiyama Y, Nagura K, Komatsu Y (2013) Novel humanized anti-CD20 antibody BM-ca binds to a unique epitope and exerts stronger cellular activity than others. Cancer Med 2:130–143
Tuscano JM, Ma Y, Martin SM, Kato J, O’Donnell RT (2011) The Bs20x22 anti-CD20-CD22 bispecific antibody has more lymphomacidal activity than do the parent antibodies alone. Cancer Immunol Immunother 60:771–780
Zhao L, Tong Q, Qian W, Li B, Zhang D, Fu T, Duan S, Zhang X, Zhao J, Dai J, Wang H, Hou S, Guo Y (2013) Eradication of non-Hodgkin lymphoma through the induction of tumor-specific T-cell immunity by CD20-Flex BiFP. Blood 122:4230–4236
Piccione EC, Juarez S, Liu J, Tseng S, Ryan C, Narayanan C, Wang L, Weiskopf K, Majeti R (2015) A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs. doi:10.1080/19420862.2015.1062192
Ruf P, Lindhofer H (2001) Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood 98:2526–2534
Stanglmaier M, Faltin M, Ruf P, Bodenhausen A, Schroder P, Lindhofer H (2008) Bi20 (fBTA05), a novel trifunctional bispecific antibody (anti-CD20 x anti-CD3), mediates efficient killing of B-cell lymphoma cells even with very low CD20 expression levels. Int J Cancer 123:1181–1189
Damm JK, Gordon S, Ehinger M, Jerkeman M, Gullberg U, Hultquist A, Drott K (2015) Pharmacologically relevant doses of valproate upregulate CD20 expression in three diffuse large B-cell lymphoma patients in vivo. Exp Hematol Oncol 4:4
Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, Yan X, Clark R, Reyes A, Stefanich E, Mai E, Young J, Johnson C, Huseni M, Wang X, Chen Y, Wang P, Wang H, Dybdal N, Chu YW, Chiorazzi N, Scheer JM, Junttila T, Totpal K, Dennis MS, Ebens AJ (2015) Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med 7:287ra70
Jakob CG, Edalji R, Judge RA, DiGiammarino E, Li Y, Gu J, Ghayur T (2013) Structure reveals function of the dual variable domain immunoglobulin (DVD-Ig) molecule. MAbs 5:358–363
Zeng J, Liu R, Wang J, Fang Y (2015) A bispecific antibody directly induces lymphoma cell death by simultaneously targeting CD20 and HLA-DR. J Cancer Res Clin Oncol. doi:10.1007/s00432-015-1949-7
Gupta P, Goldenberg DM, Rossi EA, Cardillo TM, Byrd JC, Muthusamy N, Furman RR, Chang CH (2012) Dual-targeting immunotherapy of lymphoma: potent cytotoxicity of anti-CD20/CD74 bispecific antibodies in mantle cell and other lymphomas. Blood 119:3767–3778
Fisher RI, Kaminski MS, Wahl RL, Knox SJ, Zelenetz AD, Vose JM, Leonard JP, Kroll S, Goldsmith SJ, Coleman M (2005) Tositumomab and iodine-131 tositumomab produces durable complete remissions in a subset of heavily pretreated patients with low-grade and transformed non-Hodgkin’s lymphomas. J Clin Oncol 23:7565–7573
Jacobs SA (2007) Yttrium ibritumomab tiuxetan in the treatment of non-Hodgkin’s lymphoma: current status and future prospects. Biologics 1:215–227
Pandey U, Kameswaran M, Dev SH, Samuel G (2014) 99mTc carbonyl DTPA-rituximab: preparation and preliminary bioevaluation. Appl Radiat Isot 86:52–56
Sharkey RM, Karacay H, Litwin S, Rossi EA, McBride WJ, Chang CH, Goldenberg DM (2008) Improved therapeutic results by pretargeted radioimmunotherapy of non-Hodgkin’s lymphoma with a new recombinant, trivalent, anti-CD20, bispecific antibody. Cancer Res 68:5282–5290
Law CL, Cerveny CG, Gordon KA, Klussman K, Mixan BJ, Chace DF, Meyer DL, Doronina SO, Siegall CB, Francisco JA, Senter PD, Wahl AF (2004) Efficient elimination of B-lineage lymphomas by anti-CD20-auristatin conjugates. Clin Cancer Res 10:7842–7851
Li ZH, Zhang Q, Wang HB, Zhang YN, Ding D, Pan LQ, Miao D, Xu S, Zhang C, Luo PH, Naranmandura H, Chen SQ (2014) Preclinical studies of targeted therapies for CD20-positive B lymphoid malignancies by Ofatumumab conjugated with auristatin. Invest New Drugs 32:75–86
Polito L, Bolognesi A, Tazzari PL, Farini V, Lubelli C, Zinzani PL, Ricci F, Stirpe F (2004) The conjugate Rituximab/saporin-S6 completely inhibits clonogenic growth of CD20-expressing cells and produces a synergistic toxic effect with Fludarabine. Leukemia 18:1215–1222
Vincent M, Teppaz G, Lajoie L, Sole V, Bessard A, Maillasson M, Loisel S, Bechard D, Clemenceau B, Thibault G, Garrigue-Antar L, Jacques Y, Quemener A (2014) Highly potent anti-CD20-RLI immunocytokine targeting established human B lymphoma in SCID mouse. MAbs 6:1026–1037
Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH (2009) CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood 114:3864–3871
Rossi EA, Rossi DL, Stein R, Goldenberg DM, Chang CH (2010) A bispecific antibody-IFNalpha2b immunocytokine targeting CD20 and HLA-DR is highly toxic to human lymphoma and multiple myeloma cells. Cancer Res 70:7600–7609
Marusic C, Novelli F, Salzano AM, Scaloni A, Benvenuto E, Pioli C, Donini M (2015) Production of an active anti-CD20-hIL-2 immunocytokine in Nicotiana benthamiana. Plant Biotechnol J. doi:10.1111/pbi.12378
Ocampo García BE, Miranda Olvera RM, Santos Cuevas CL, García Becerra R, Azorín Vega EP, Ordaz Rosado D (2014) In vitro decrease of the BCL-2 protein levels in lymphoma cells induced by gold nanoparticles and gold nanoparticles-anti-CD20. Nanosci Technol 1:1–6
Minai L, Yeheskely-Hayon D, Yelin D (2013) High levels of reactive oxygen species in gold nanoparticle-targeted cancer cells following femtosecond pulse irradiation. Sci Rep 3:2146
Voltan R, Secchiero P, Ruozi B, Forni F, Agostinis C, Caruso L, Vandelli MA, Zauli G (2013) Nanoparticles engineered with rituximab and loaded with Nutlin-3 show promising therapeutic activity in B-leukemic xenografts. Clin Cancer Res 19:3871–3880
Sapra P, Allen TM (2004) Improved outcome when B-cell lymphoma is treated with combinations of immunoliposomal anticancer drugs targeted to both the CD19 and CD20 epitopes. Clin Cancer Res 10:2530–2537
Davidson N, Camburn T, Keary I, Houghton D (2014) Substituting Doxorubicin with nonpegylated liposomal Doxorubicin for the treatment of early breast cancer: results of a retrospective study. Int J Breast Cancer 2014:984067
Iannitto E, Luminari S, Tripodo C, Mancuso S, Cesaretti M, Marcheselli L, Merli F, Stelitano C, Carella AM, Fragasso A, Montechiarello E, Ricciuti G, Pulsoni A, Paulli M, Franco V, Federico M (2015) Rituximab with cyclophosphamide, vincristine, non-pegylated liposomal doxorubicin and prednisone as first-line treatment for splenic marginal zone lymphoma: a Fondazione Italiana Linfomi phase II study. Leuk Lymphoma. doi:10.3109/10428194.2015.1029925
Yu B, Mao Y, Bai LY, Herman SE, Wang X, Ramanunni A, Jin Y, Mo X, Cheney C, Chan KK, Jarjoura D, Marcucci G, Lee RJ, Byrd JC, Lee LJ, Muthusamy N (2013) Targeted nanoparticle delivery overcomes off-target immunostimulatory effects of oligonucleotides and improves therapeutic efficacy in chronic lymphocytic leukemia. Blood 121:136–147
Chu TW, Yang J, Kopecek J (2012) Anti-CD20 multivalent HPMA copolymer-Fab’ conjugates for the direct induction of apoptosis. Biomaterials 33:7174–7181
Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ (2010) Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant 16:1245–1256
Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, Gopal AK, Pagel JM, Lindgren CG, Greenberg PD, Riddell SR, Press OW (2008) Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 112:2261–2271
Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, Raubitschek A, Forman SJ, Greenberg PD, Riddell SR, Press OW (2012) CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 119:3940–3950
Jensen M, Tan G, Forman S, Wu AM, Raubitschek A (1998) CD20 is a molecular target for scFvFc:zeta receptor redirected T cells: implications for cellular immunotherapy of CD20+ malignancy. Biol Blood Marrow Transplant 4:75–83
Jensen MC, Cooper LJ, Wu AM, Forman SJ, Raubitschek A (2003) Engineered CD20-specific primary human cytotoxic T lymphocytes for targeting B-cell malignancy. Cytotherapy 5:131–138
Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, Till B, Raubitschek A, Forman SJ, Qian X, James S, Greenberg P, Riddell S, Press OW (2007) Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther 18:712–725
Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, Brouns SA, Spencer DM, Till BG, Jensen MC, Riddell SR, Press OW (2013) Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One 8, e82742
Muller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, Tonn T, Wels WS (2008) Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother 57:411–423
Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, Klingemann H (2013) Retargeting NK-92 cells by means of. Oncoimmunology 2, e26527
Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS (2015) Targeting CD20+ aggressive B-cell non-hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res 3:333–344
Reagan MR, Seib FP, McMillin DW, Sage EK, Mitsiades CS, Janes SM, Ghobrial IM, Kaplan DL (2012) Stem cell implants for cancer therapy: TRAIL-expressing Mesenchymal stem cells target cancer cells in situ. J Breast Cancer 15:273–282
Yan C, Li S, Li Z, Peng H, Yuan X, Jiang L, Zhang Y, Fan D, Hu X, Yang M, Xiong D (2013) Human umbilical cord mesenchymal stem cells as vehicles of CD20-specific TRAIL fusion protein delivery: a double-target therapy against non-Hodgkin’s lymphoma. Mol Pharm 10:142–151
Bremer E, Ten CB, Samplonius DF, Mueller N, Wajant H, Stel AJ, Chamuleau M, van de Loosdrecht AA, Stieglmaier J, Fey GH, Helfrich W (2008) Superior activity of fusion protein scFvRit:sFasL over cotreatment with rituximab and Fas agonists. Cancer Res 68:597–604
Brunekreeft KL, Strohm C, Gooden MJ, Rybczynska AA, Nijman HW, Grigoleit GU, Helfrich W, Bremer E, Siegmund D, Wajant H, de Bruyn M (2014) Targeted delivery of CD40L promotes restricted activation of antigen-presenting cells and induction of cancer cell death. Mol Cancer 13:85
Gisselbrecht C, Schmitz N, Mounier N, Singh GD, Linch DC, Trneny M, Bosly A, Milpied NJ, Radford J, Ketterer N, Shpilberg O, Duhrsen U, Hagberg H, Ma DD, Viardot A, Lowenthal R, Briere J, Salles G, Moskowitz CH, Glass B (2012) Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol 30:4462–4469
Abou-Nassar KE, Stevenson KE, Antin JH, McDermott K, Ho VT, Cutler CS, Lacasce AS, Jacobsen ED, Fisher DC, Soiffer RJ, Alyea EP, Koreth J, Freedman AS (2011) (90)Y-ibritumomab tiuxetan followed by reduced-intensity conditioning and allo-SCT in patients with advanced follicular lymphoma. Bone Marrow Transplant 46:1503–1509
Chang JE, Seo S, Kim KM, Werndli JE, Bottner WA, Rodrigues GA, Sanchez FA, Saphner TJ, Longo WL, Kahl BS (2010) Rituximab and CHOP chemotherapy plus GM-CSF for previously untreated diffuse large B-cell lymphoma in the elderly: a Wisconsin oncology network study. Clin Lymphoma Myeloma Leuk 10:379–384
Schliemann C, Palumbo A, Zuberbuhler K, Villa A, Kaspar M, Trachsel E, Klapper W, Menssen HD, Neri D (2009) Complete eradication of human B-cell lymphoma xenografts using rituximab in combination with the immunocytokine L19-IL2. Blood 113:2275–2283
Rosario M, Liu B, Kong L, Schneider SE, Jeng EK, Rhode PR, Wong H, Fehniger TA (2014) The IL-15 superagonist ALT-803 enhances NK cell ADCC and in vivo clearance of B cell lymphomas directed by an anti-CD20 monoclonal antibody. Blood 124:807
Timmerman JM, Byrd JC, Andorsky DJ, Yamada RE, Kramer J, Muthusamy N, Hunder N, Pagel JM (2012) A phase I dose-finding trial of recombinant interleukin-21 and rituximab in relapsed and refractory low grade B-cell lymphoproliferative disorders. Clin Cancer Res 18:5752–5760
Abes R, Gelize E, Fridman WH, Teillaud JL (2010) Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 116:926–934
Manzur S, Cohen S, Haimovich J, Hollander N (2012) Enhanced therapeutic effect of B cell-depleting anti-CD20 antibodies upon combination with in-situ dendritic cell vaccination in advanced lymphoma. Clin Exp Immunol 170:291–299
Oksvold MP, Kullmann A, Forfang L, Kierulf B, Li M, Brech A, Vlassov AV, Smeland EB, Neurauter A, Pedersen KW (2014) Expression of B-cell surface antigens in subpopulations of exosomes released from B-cell lymphoma cells. Clin Ther 36:847–862
Rialland P, Lankar D, Raposo G, Bonnerot C, Hubert P (2006) BCR-bound antigen is targeted to exosomes in human follicular lymphoma B-cells. Biol Cell 98:491–501
Bennit HRF, Valenzuela MMA, Jutzy JS, Wall NP (2014) B-cell lymphoma-derived exosomes are reservoirs of inhibitors of apoptosis. Cancer Res 74:1108
Aung T, Chapuy B, Vogel D, Wenzel D, Oppermann M, Lahmann M, Weinhage T, Menck K, Hupfeld T, Koch R, Trumper L, Wulf GG (2011) Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc Natl Acad Sci U S A 108:15336–15341
Stuhmer T, Zollinger A, Siegmund D, Chatterjee M, Grella E, Knop S, Kortum M, Unzicker C, Jensen MR, Quadt C, Chene P, Schoepfer J, Garcia-Echeverria C, Einsele H, Wajant H, Bargou RC (2008) Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 22:1604–1612
Richter K, Muschler P, Hainzl O, Reinstein J, Buchner J (2003) Sti1 is a non-competitive inhibitor of the Hsp90 ATPase. Binding prevents the N-terminal dimerization reaction during the atpase cycle. J Biol Chem 278:10328–10333
Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, Knight J, Starostik P, Deans J, Hernandez-Ilizaliturri FJ (2008) Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res 14:1561–1570
Ottosson-Wadlund A, Ceder R, Preta G, Pokrovskaja K, Grafstrom RC, Heyman M, Soderhall S, Grander D, Hedenfalk I, Robertson JD, Fadeel B (2013) Requirement of apoptotic protease-activating factor-1 for bortezomib-induced apoptosis but not for Fas-mediated apoptosis in human leukemic cells. Mol Pharmacol 83:245–255
Gressin R, Houot R, Uribe MO, Mounier C, Bouabdallah K, Alexis M, Senecal D, Molles MP, Tournilhac O, Park S, Rodon P, Yamani AEI, Sutton L, Lioure B, Assouline D, Harousseau J, Maisonneuve H, Caulet S, Gouill SL (2010) Final results of the RiPAD+C regimen including Velcade in front line therapy for elderly patients with mantle cell lymphoma. A phase II prospective study of the GOELAMS group. Blood 116:2791
Ruan J, Martin P, Furman RR, Lee SM, Cheung K, Vose JM, Lacasce A, Morrison J, Elstrom R, Ely S, Chadburn A, Cesarman E, Coleman M, Leonard JP (2011) Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 29:690–697
Liu H, Westergard TD, Cashen A, Piwnica-Worms DR, Kunkle L, Vij R, Pham CG, DiPersio J, Cheng EH, Hsieh JJ (2014) Proteasome inhibitors evoke latent tumor suppression programs in pro-B MLL leukemias through MLL-AF4. Cancer Cell 25:530–542
Riaz W, Hernandez-Ilizaliturri FJ, Mavis C, Tsai P, Czuczman MS (2010) Efficacy of combination of rituximab (R), obatoclax (O), and bortezomib (B) against rituximab-sensitive (RSCL) and rituximab-resistant B-cell lymphoma cell lines (RRCL). J Clin Oncol 28:8094
Johnson AJ, Wagner AJ, Cheney CM, Smith LL, Lucas DM, Guster SK, Grever MR, Lin TS, Byrd JC (2007) Rituximab and 17-allylamino-17-demethoxygeldanamycin induce synergistic apoptosis in B-cell chronic lymphocytic leukaemia. Br J Haematol 139:837–844
Reddy N, Hicks D, Jagasia M, Amiri K (2009) SNX 2112, an oral Hsp-90 inhibitor exerts antiproliferative effects in combination with bortezomib and rituximab in rituximab resistant non-Hodgkin’s lymphoma. Blood 114:3733
Ashkenazi A, Holland P, Eckhardt SG (2008) Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol 26:3621–3630
Maddipatla S, Hernandez-Ilizaliturri FJ, Knight J, Czuczman MS (2007) Augmented antitumor activity against B-cell lymphoma by a combination of monoclonal antibodies targeting TRAIL-R1 and CD20. Clin Cancer Res 13:4556–4564
Yee L, Fanale M, Dimick K, Calvert S, Robins C, Ing J, Novotny W, Ashkenazi A, Burris IH (2007) A phase IB safety and pharmacokinetic (PK) study of recombinant human Apo2L/TRAIL in combination with rituximab in patients with low-grade non-Hodgkin lymphoma. J Clin Oncol 25:8078
Cheah CY, Belada D, Fanale MA, Janikova A, Czucman MS, Flinn IW, Kapp AV, Ashkenazi A, Kelley S, Bray GL, Holden S, Seymour JF (2015) Dulanermin with rituximab in patients with relapsed indolent B-cell lymphoma: an open-label phase 1b/2 randomised study. Lancet Haematol 2:166–174
Stephens DM, Ruppert AS, Maddocks K, Andritsos L, Baiocchi R, Jones J, Johnson AJ, Smith LL, Zhao Y, Ling Y, Li J, Phelps MA, Grever MR, Byrd JC, Flynn JM (2013) Cyclophosphamide, alvocidib (flavopiridol), and rituximab, a novel feasible chemoimmunotherapy regimen for patients with high-risk chronic lymphocytic leukemia. Leuk Res 37:1195–1199
Fabre C, Gobbi M, Ezzili C, Zoubir M, Sablin MP, Small K, Im E, Shinwari N, Zhang D, Zhou H, Le TC (2014) Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients. Cancer Chemother Pharmacol 74:1057–1064
Seki R, Ohshima K, Fujisaki T, Uike N, Kawano F, Gondo H, Makino S, Eto T, Moriuchi Y, Taguchi F, Kamimura T, Tsuda H, Shimoda K, Okamura T (2010) Prognostic significance of S-phase kinase-associated protein 2 and p27kip1 in patients with diffuse large B-cell lymphoma: effects of rituximab. Ann Oncol 21:833–841
Mahadevan D, Stejskal A, Cooke LS, Manziello A, Morales C, Persky DO, Fisher RI, Miller TP, Qi W (2012) Aurora A inhibitor (MLN8237) plus vincristine plus rituximab is synthetic lethal and a potential curative therapy in aggressive B-cell non-Hodgkin lymphoma. Clin Cancer Res 18:2210–2219
Xu ZZ, Wang WF, Fu WB, Wang AH, Liu ZY, Chen LY, Guo P, Li JM (2014) Combination of rituximab and mammalian target of rapamycin inhibitor everolimus (RAD001) in diffuse large B-cell lymphoma. Leuk Lymphoma 55:1151–1157
Prasad A, Shrivastava A, Papadopoulos E, Kuzontkoski PM, Reddy MV, Gillum AM, Kumar R, Reddy EP, Groopman JE (2013) Combined administration of rituximab and on 013105 induces apoptosis in mantle cell lymphoma cells and reduces tumor burden in a mouse model of mantle cell lymphoma. Clin Cancer Res 19:85–95
Wang ML, Lee H, Chuang H, Wagner-Bartak N, Hagemeister F, Westin J, Fayad L, Samaniego F, Turturro F, Oki Y, Chen W, Badillo M, Nomie K, Rosa MD, Zhao D, Lam L, Addison A, Zhang H, Young KH, Li S, Santos D, Medeiros LJ, Champlin R, Romaguera J, Zhang L (2016) Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol 17:48–56
O’Brien SM, Lamanna N, Kipps TJ, Flinn I, Zelenetz AD, Burger JA, Keating M, Mitra S, Holes L, Yu AS, Johnson DM, Miller LL, Kim Y, Dansey RD, Dubowy RL, Coutre SE (2015) A phase 2 study of idelalisib plus rituximab in treatment-naïve older patients with chronic lymphocytic leukemia. Blood 17(126):2686–2694
Zhang LH, Kosek J, Wang M, Heise C, Schafer PH, Chopra R (2013) Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br J Haematol 160:487–502
Fecteau JF, Corral LG, Ghia EM, Gaidarova S, Futalan D, Bharati IS, Cathers B, Schwaederle M, Cui B, Lopez-Girona A, Messmer D, Kipps TJ (2014) Lenalidomide inhibits the proliferation of CLL cells via a cereblon/p21(WAF1/Cip1)-dependent mechanism independent of functional p53. Blood 124:1637–1644
Hernandez-Ilizaliturri FJ, Reddy N, Holkova B, Ottman E, Czuczman MS (2005) Immunomodulatory drug CC-5013 or CC-4047 and rituximab enhance antitumor activity in a severe combined immunodeficient mouse lymphoma model. Clin Cancer Res 11:5984–5992
Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, Roth M, Vaughn M, Knight J, Wallace P, Czuczman MS (2008) Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol 140:36–45
Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, Schafer P, Bartlett JB (2008) lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res 14:4650–4657
Gaidarova S, Corral L, Glezer E, Schafer P, Lopez-Girona A (2009) Treatment of MCL cells with combined rituximab and lenalidomide enhances NK-mediated synapse formation and cell killing. ASH Ann Meet 114:1687
Gaidarova S, Mendy D, Heise C, Aukerman S, Daniel T, Chopra R, Lopez-Girona A (2010) Lenalidomide induces capping of CD20 and cytoskeleton proteins to enhance rituximab immune recognition of malignant B-cells. ASH Annual Meeting 116:2845
Fowler NH, Davis RE, Rawal S, Nastoupil L, Hagemeister FB, McLaughlin P, Kwak LW, Romaguera JE, Fanale MA, Fayad LE, Westin JR, Shah J, Orlowski RZ, Wang M, Turturro F, Oki Y, Claret LC, Feng L, Baladandayuthapani V, Muzzafar T, Tsai KY, Samaniego F, Neelapu SS (2014) Safety and activity of lenalidomide and rituximab in untreated indolent lymphoma: an open-label, phase 2 trial. Lancet Oncol 15:1311–1318
Chong EA, Ahmadi T, Aqui NA, Svoboda J, Nasta SD, Mato AR, Walsh KM, Schuster SJ (2015) Combination of Lenalidomide and rituximab overcomes rituximab resistance in patients with indolent B-cell and mantle cell lymphomas. Clin Cancer Res 21:1835–1842
Ruan J, Martin P, Shah BD, Schuster SJ, Smith SM, Furman RR, Christos P, Rodriguez A, Wolstencroft P, Svoboda J, Bender A, Lewis J, Coleman M, Leonard JP (2014) Sustained remission with the combination biologic doublet of lenalidomide plus rituximab as initial treatment for mantle cell lymphoma: a multi-center phase II study report. Blood 124:625
Flinn I, Mainwaring M, Peacock N, Shipley D, Arrowsmith E, Savona M, Hainsworth JD, Berdeja JG (2012) Rituximab, lenalidomide, and bortezomib in the first-line or second-line treatment of patients with mantle cell lymphoma a phase I/II trial. ASH Annual Meeting 120:2748
Hagner PR, Man HW, Fontanillo C, Wang M, Couto S, Breider M, Bjorklund C, Havens CG, Lu G, Rychak E, Raymon H, Narla RK, Barnes L, Khambatta G, Chiu H, Kosek J, Kang J, Amantangelo MD, Waldman M, Lopez-Girona A, Cai T, Pourdehnad M, Trotter M, Daniel TO, Schafer PH, Klippel A, Thakurta A, Chopra R, Gandhi AK (2015) CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood. doi:10.1182/blood-2015-02-628669
Hasanali Z, Sharma K, Spurgeon S, Okada C, Stuart A, Shimko S, Leshchenko V, Parekh S, Chen Y, Kirschbaum M, Epner EM (2013) Combined epigenetic and immunotherapy produces dramatic responses in 100% of newly diagnosed mantle cell lymphoma patients. Cancer Res 73:LB-140
Sharma K, Leshchenko VV, Hasanali Z, Stuart A, Shimko S, Spurgeon SE, Parekh S, Epner EM (2013) Combined epigenetic and immunotherapy for newly diagnosed mantle cell lymphoma: correlative studies suggest the importance of enhanced ADCC, mechanisms of resistance and cyclin D1 nuclear localization genotype. Blood 122:3063
Spurgeon S, Sharma K, Claxton DF, Ehmann C, Shimko S, Stewart A, Parekh S, Leshchenko VV, Chen Y, Mori M, Pu JJ, Epner EM (2014) Final results of a phase 1–2 study of vorinostat (SAHA), cladribine, and rituximab (SCR) relapsed B-Cell non-Hodgkin’s lymphoma and previously untreated mantle cell lymphoma. Blood 124:1714
Spurgeon S, Chen AI, Okada C, Parekh S, Leshchenko VV, Palmbach G, Ratterree B, Subbiah N, Capper C, Epner EM (2011) A Phase I/II Study ofvorinostat (SAHA), cladribine (2-CdA), and rituximab shows significant activity inpreviously untreated mantle cell lymphoma. Blood 118:203
Chen R, Frankel P, Popplewell L, Siddiqi T, Ruel N, Rotter A, Thomas SH, Mott M, Nathwani N, Htut M, Nademanee A, Forman SJ, Kirschbaum M (2015) A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica 100:357–362
Gopal AK, Pagel JM, Hedin N, Press OW (2004) Fenretinide enhances rituximab-induced cytotoxicity against B-cell lymphoma xenografts through a caspase-dependent mechanism. Blood 103:3516–3520
Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP (2004) PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res 64:5471–5480
Pagel JM, Laugen C, Bonham L, Hackman RC, Hockenbery DM, Bhatt R, Hollenback D, Carew H, Singer JW, Press OW (2005) Induction of apoptosis using inhibitors of lysophosphatidic acid acyltransferase-beta and anti-CD20 monoclonal antibodies for treatment of human non-Hodgkin's lymphomas. Clin Cancer Res 11:4857–4866
Winiarska M, Nowis D, Bil J, Glodkowska-Mrowka E, Muchowicz A, Wanczyk M, Bojarczuk K, Dwojak M, Firczuk M, Wilczek E, Wachowska M, Roszczenko K, Miaczynska M, Chlebowska J, Basak GW, Golab J (2012) Prenyltransferases regulate CD20 protein levels and influence anti-CD20 monoclonal antibody-mediated activation of complement-dependent cytotoxicity. J Biol Chem 287:31983–31993
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Rights and permissions
About this article

Cite this article
Safdari, Y., Ahmadzadeh, V. & Farajnia, S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs 34, 497–512 (2016). https://doi.org/10.1007/s10637-016-0349-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-016-0349-4