
Abstract
For decades, the pathogenesis of a variety of human diseases has been attributed to increased intestinal paracellular permeability even though scientific evidence supporting this hypothesis has been tenuous. Nevertheless, during the past decade, there have been a growing number of publications focused on human genetics, the gut microbiome, and proteomics, suggesting that loss of mucosal barrier function, particularly in the gastrointestinal tract, may substantially affect antigen trafficking, ultimately causing chronic inflammation, including autoimmunity, in genetically predisposed individuals. The gut mucosa works as a semipermeable barrier in that it permits nutrient absorption and also regulates immune surveillance while retaining potentially harmful microbes and environmental antigens within the intestinal lumen. Celiac disease (CD), a systemic, immune-mediated disorder triggered by gluten in genetically susceptible individuals, is associated with altered gut permeability. Pre-clinical and clinical studies have shown that gliadin, a prolamine component of gluten that is implicated in CD pathogenesis, is capable to disassembling intercellular junctional proteins by upregulating the zonulin pathway, which can be inhibited by the zonulin antagonist larazotide acetate. In this review, we will focus on CD as a paradigm of chronic inflammatory diseases in order to outline the contribution of gut paracellular permeability toward disease pathogenesis; moreover, we will summarize current evidence derived from available clinical trials of larazotide acetate in CD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Celiac disease (CD) is a systemic, immune-mediated disorder triggered by gluten and related prolamines (such as gliadin) in genetically susceptible individuals that encompasses the presence of a variable combination of clinical manifestations, CD-specific antibodies, human leukocyte antigen (HLA)-DQ2 or HLA-DQ8 haplotypes, and immune-mediated enteropathy [1]. Although CD may manifest at any age [2], a majority of subjects develops the disease in their first five years of life [3]. Given its mutable blend of systemic and malabsorptive clinical features, CD has been likened to a “tricky clinical chameleon” for primary care physicians [4]. Nowadays, many patients with CD have a paucity of symptoms or primarily display extraintestinal symptoms, whereas only a minority has the “classical” clinical picture of malabsorption with weight loss/failure to thrive and chronic diarrhea [5].
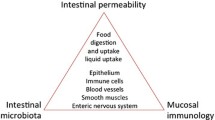
In line with the increasing prevalence of many chronic inflammatory and autoimmune diseases [6], CD prevalence has been on the rise worldwide during the last 4–5 decades [7,8,9]. The causes underlying this epidemiologic phenomenon have been historically linked to the hygiene hypothesis, which posits that increased hygienic practice has decreased early life exposure to pathogens that are essential for the normal development of the immune system [10]. With increased knowledge on the pathogenesis of chronic inflammatory diseases, gut barrier dysfunction, aberrant immunity, and an imbalanced microbiome composition (namely dysbiosis) have been identified as essential contributors to the pathogenesis of these conditions, including CD [11] (Fig. 1).

Modern system biology pathogenic theory of chronic inflammatory diseases. The fragile balance between health and disease is determined by several factors such as genetic background, exposure to environmental factors, loss of gut barrier function, inappropriate mucosal immunity, and microbiome imbalance. The onset and the natural history of several diseases are the outcome of changes of these tightly interlaced and mutually influencing elements. Predicting models of health according to these factors are the ultimate goals of primary prevention in the boosting field of “personalized precision” medicine
In this review, we will focus on CD as a paradigm of a chronic inflammatory disease in order to better understand the relation of gut permeability to disease pathogenesis.
Structure and Function of the Intestinal Barrier
The gut mucosa encompasses the largest interface between the human body and the external environment, with numerous implications for health and disease (Fig. 2) [12]. The gut mucosa acts as a semipermeable fence: It permits nutrient transport and immune detection, while strongly restricting the passage of potentially harmful microbes and environmental antigens from the lumen to the systemic circulation. This physical barrier is composed of the mucus layer, the glycocalyx on the luminal surface of the intestinal epithelial cells (IEC), and the intercellular junctions dynamically connecting neighborhood intestinal epithelial cells (IEC) [13].

Proposed involvement of barrier dyfunction in the pathogenesis of non-infective chronic inflammatory diseases. (1) Under physiological circumstances, there is a tightly control of antigen trafficking from the gut lumen into the intestinal lamina propria (antigen sampling) that leads to mucosal tolerance, homeostasis and anergy mediated by the involvement of a variety immune cells and anti-inflammatory cytokines. (2) Following a variety of environmental stimuli, including gut microbiome imbalance (dysbiosis), paracellular permeability is increased secondary to the upregulation of specific pathways, including the zonulin pathway. (3) As a consequence of prolonged and uncontrolled antigen trafficking, innate immune response or immunoregulatory defects leads to the onset of inflammation with secondary production of pro-inflammatory cutokines, including TNF-aloha and IFN-gamma that, by themselves, are causing further increase in gut permeability by activating the myosin light chain kinake (MLCK) pathway. (4) This mechanism starts a vicious cycle of further increase in gut permeability and subsequent antigen trafficking that ultimately lead to break of tolerance and onset of chronci inflammatory diseases whose nature depends on the genetic host predisposition
The mucus layer, secreted by goblet cells residing within the mucosa, is composed by mucin glycoproteins that serve many functions, one being impeding the entry of microorganisms into the submucosa [14]. Similar to the mucus layer, the glycocalyx represents an interlaced structure of carbohydrate and glycolipids or glycoproteins that serve as a secondary hurdle for pathogens prior to reaching the epithelial cells [15]. In health, the intestinal epithelial layer is impermeable to hydrophilic solutes lacking specific transporters. Each epithelial cell is linked to its neighbor cell by junctional proteins, thus preventing uncontrolled paracellular transport of large molecules. Three molecular complexes constitute this junctional system: tight junctions (TJs), adherens junctions (AJs), and desmosomes [16].
Tight junctions are the most apical junctional complex, creating a polarized system separating the apical and basolateral cellular poles. It was previously believed that tight junctions were impermeable and static, thus forming a fixed barrier. In 1981, this paradigm was upended by the finding that cyclic adenosine monophosphates altered the permeability of gallbladder epithelial tight junctions [17]. Following this discovery, there was still much debate regarding the mechanisms by which paracellular permeability was regulated with the structure of tight junctions unknown. Finally, in 1993 the first tight junction-associated protein, zonula occludens (ZO)-1, was discovered [18]. Currently, the consensus is that several transmembrane proteins are assembled to form tight junctions: occludin [19], claudins [20], junctional adhesion molecules (JAMs) [21], tricellulin [22], and angulins [23]. TJ development relies on AJ formation, as ZO-1 protein drifts apically toward occludin only after AJ assembly [24].
CD Pathogenesis
CD is a unique autoimmune disease since its key genetic elements (HLA-DQ2 and HLA-DQ8), the auto-antigen involved (tissue transglutaminase 2: tTG2), and the environmental trigger (gluten) are all well defined. As HLA-DQ2/HLA-DQ8 is frequent among the general population (25–35%) with only 3% of these HLA-compatible individuals developing CD [25], it is not surprising that genome-wide association studies (GWASs) have identified more than 100 non-HLA genes associated with CD [26, 27]. Although the relevance of these additional risk genes for CD is rather limited, they provide key clues to the pathways potentially involved in disease pathogenesis. Affecting only humans, a major drawback in CD research has been the lack of a reliable and reproducible animal model. Nevertheless, new technologies pertinent to human gut biology and immunology such as gut organoids are opening unprecedented opportunities for major research breakthroughs [28]. As with many other autoimmune diseases, an epidemic of CD has occurred, questioning the long-held paradigm that genetic predisposition and gluten ingestion are necessary and sufficient elements for disease manifestation.
During the past 40 years, improved hygiene and lack of exposure to microorganisms have also been linked to a steep increase in autoimmune disorders in industrialized countries [29]. The hygiene hypothesis argues that the rising incidence of many autoimmune diseases may partially be the result of lifestyle and environmental changes that have made humans in developed countries too “clean” for their own good. With breakthroughs in the understanding of how the gut microbiological ecosystem balances tolerance and the immune response, this hypothesis is now under scrutiny. Regardless of whether autoimmune diseases are due to too much or too little exposure to environmental microorganisms, inappropriate antigen trafficking secondary to increased gut permeability, the imbalance in T helper cell responses, and gut dysbiosis are key elements of the pathogenesis of the autoimmune process [30].
Besides genetic predisposition and exposure to gluten, loss of intestinal barrier function and a proinflammatory innate immune response triggered by gluten and by an imbalanced gut microbiome all seem to be pivotal cofactors preceding the CD-related autoimmune “storm” [31,32,33].
CD Pathogenesis: The Zonulin Pathway as Master Regulator of Gut Permeability
Identification of Pre-haptoglobin 2 as the Archetype of the Zonulin Family
Through proteomic analysis of CD patients sera, the first member of the zonulin family was identified as pre-haptoglobin (HP)2 [34], described as the inactive precursor of HP2, one of the two genetic variants (together with HP1) of human HPs. Mature human HPs are plasma glycoproteins composed of α and β polypeptide chains that are covalently associated by disulfide bonds. While the β chain (36 kDa) is constant, the α chain exists in two isoforms, i.e., α1 (~ 9 kDa) and α2 (~ 18 kDa). The presence of one or both of the α chains produces the three human HP phenotypes (HP1-1 homozygote, HP2-1 heterozygote, and HP2-2 homozygote). Despite this multidomain structure, the only function assigned to HPs to date is to bind hemoglobin (Hb) to form stable HP-Hb complexes, thereby preventing Hb-induced oxidative tissue damage [35]. In contrast, no function has ever been described for their precursor forms. The primary translation product of mammalian HP mRNA is a polypeptide that dimerizes co-translationally and is proteolytically cleaved, while it is still in the endoplasmic reticulum [36]. Conversely, zonulin is detectable in an uncleaved form in human serum, adding another intriguing aspect to the multifunctional characteristics of HPs. HPs are unusual secretory proteins in that they are proteolytically processed in the endoplasmic reticulum, the subcellular fraction that contains the highest zonulin concentration [37].
Structural Characterization of Zonulin Archetype and Its Subunits
Phylogenetic analyses suggest that HPs evolved from a complement-associated protein (mannose-binding lectin-associated serine protease, MASP), with their α chain containing a complement control protein (CCP; the domain that activates complement), whereas the β chain is related to chymotrypsin-like serine proteases (SP domain) [38, 39]. Nevertheless, the SP domain of HP lacks the essential catalytic amino acid residues required for protease function; recent structure–function analyses have implicated this domain in receptor recognition and binding [40, 41].
Although not a serine protease, zonulin shares about 19% amino acid sequence homology with chymotrypsin; their genes both map to chromosome 16. The active site residues typical of the serine proteases, histidine-57 and serine-195, are replaced in zonulin by lysine and alanine, respectively. Due to these mutations, zonulin most likely lost its protease activity during evolution, even though zonulin and serine proteases evolved from a common ancestor [38]. Therefore, zonulin and the serine proteases represent a striking example of homologous proteins with different biological functions. Other members of the MASP family include a series of plasminogen-related growth factors such as epidermal growth factor (EGF), hepatocyte growth factor (HGF), and others factors involved in cell growth, proliferation, differentiation, migration, and disruption of intercellular junctions. Another element supporting the fact that zonulin belongs to the MASP family is that it shared the same receptor (EGF receptor, EGFR, see below) of other members related to the plasminogen-related growth factors [34].
Evolutionary and Structural Biology of the Zonulin Family: Properdin as a Second Member of the Family
The α2 chain gene (and, therefore, zonulin), found only in humans, originated approximately 2 million years ago, 500,000 years before the human and chimpanzee lineages split through a chromosomal aberration (unequal crossover) in a humanoid in India. Since our laboratory has detected zonulin in other mammals [42, 43], it is likely that frequent zonulin polymorphisms secondary to high mutation rate during evolution led to a family of structurally and functionally related zonulins. Indeed, a new member of the zonulin family have recently identified as properdin [44], another MASP protein. Once released from neutrophils, T cells, and macrophages in response to acute microbial exposure, properdin produces the chemotactic anaphylatoxin C3a and C5a with subsequent formation of immune complexes that increase endothelial permeability [45]. Intriguingly, zonulin as pre-HP2 also generates C3a and C5a, with subsequent increased vascular permeability in several organs, including the lung, with subsequent onset of acute lung injury [46]. Another striking similarity between zonulin and properdin is that both are associated with viral respiratory tract infections [47, 48]. Beside zonulin and properdin, other members of the MASP family include a series of plasminogen-related growth factors [e.g., epidermal growth factor (EGF) and (HGF)] involved in cell growth, proliferation, differentiation, and migration, as well as disruption of intercellular junctions. In light of these considerations, other MASP members identified in our capturing experiments in general, and properdin in particular, are intriguing possible targets.
Challenges in Measuring Zonulins with Currently Commercially Available ELISAs
Several groups, including ours, have been questioning what exactly the commercially available zonulin ELISA kits measure [44, 49]. Using different approaches, both we and others identified complement C3 as the top match protein identified by the antibodies used for the ELISA [44, 49]. However, complement C3 is most likely an unspecific product overshadowing the real targets. Indeed, the respective ELISA kit did not detect any complement C proteins obtained from different suppliers when tested under native and denatured conditions, as well as diluted in serum [44]. Also, the same kit did not detect recombinant zonulin, mature HP1 or mature HP2 [44]. Considering the additional mass spectrometric (MS) hits reported [44, 49], a few proteins stand out, although, without further validation, these data need to be interpreted with caution, since only more abundant proteins may be identified by MS analysis, while the protein(s) of interest belonging to the zonulin family may be in low abundance in serum samples and, therefore, not identifiable with this approach, keeping this in mind and the fact that the protein(s) of interest should be in the ~ 50 kDa range, like pre-HP2 and properdin. Combined Western blot analysis and ELISA test confirmed that the polyclonal antibodies raised against the zonulin-derived synthetic peptide AT1001 and used in the Immundiagnostik kit detect properdin among other proteins [44]. However, when purified proteins/peptides, including the AT1001 peptide used to raise the polyclonal antibodies (internal control), were tested by ELISA, they were highly underestimated by the test. One possible explanation for these results is that zonulin as both pre-HP2 and properdin are not the main targets detected by the ELISA; however, the fact that even AT1001 was underestimated seems to suggest that this hypothesis cannot entirely explain these results. Alternatively, it is possible that tertiary and quaternary (multimers) structure arrangements present in sera samples but not in recombinant proteins are necessary in order to properly detect any zonulin member by this ELISA. Based on these results, it is likely that the commercially available ELISAs detect more than one or more members of the zonulin family that have not been discovered yet.
Zonulin Signaling
Structural analysis of zonulin revealed similarities to several growth factors. Like zonulin, growth factors affect intercellular TJ integrity [50]. Our data showing that zonulin but not its cleaved subunits activates the EGFR [34] and that its effect on gut permeability was prevented by blocking the receptor suggested that zonulin is properly folded to activate EGFR and, therefore, to disassemble TJs only in its uncleaved form. Several G protein coupled receptors (GPCRs), including protease-activated receptors (PAR)2, transactivate EGFR [51]. Zonulin prokaryotic counterpart Zot active peptide FCIGRL (AT1002) has structural similarities with PAR2-activating peptide (AP), SLIGRL, that causes PAR2-dependent changes in permeability [52], a finding that we have demonstrated in wild-type (WT) but not PAR2−/− mice [34].
Stimuli That Release Zonulin in the Gut
Among the several potential intestinal luminal stimuli that can trigger zonulin release, we identified small intestinal exposure to bacteria and gluten as the two more powerful triggers. Enteric infections have been implicated in the pathogenesis of several pathological conditions, including allergic, autoimmune, and inflammatory diseases, by increasing the paracellular permeability of the intestinal barrier. We have generated evidence that the small intestine exposed to enteric bacteria secreted zonulin [42]. This secretion was independent of either the animal species from which the small intestines were isolated or the virulence of the microorganisms tested, occurred only on the luminal aspect of the bacteria-exposed small intestinal mucosa, and was followed by an increase in intestinal paracellular permeability coincident with the disengagement of the protein ZO-1 from the tight junctional complex [53]. This zonulin-driven opening of the paracellular pathway may represent a defensive mechanism, which “flushes out” microorganisms, contributing to the innate immune response of the host against bacterial colonization of the small intestine.
Besides bacterial exposure, we have shown that gliadin also affects intestinal barrier function by releasing zonulin [54]. This effect of gliadin is polarized, i.e., gliadin increases intestinal permeability only when administered on the luminal pole of the intestinal tissue [55]. Gliadins, key components of gluten, are complex proteins unusually rich in prolines and glutamines that are not completely digestible by mammalian intestinal enzymes [56]. The final product of this partial digestion is a mix of peptides that can trigger host responses (increased gut permeability and innate and adaptive immune response) that closely resemble those instigated by the exposure to potentially harmful microorganisms [57]. Moreover, gliadin immediately and transiently enhances zonulin-dependent increased gut paracellular permeability irrespective of disease status (Fig. 3). [37, 58]. This observation led us to the identification of the chemokine receptor CXCR3 as the target intestinal receptor for gliadin [32]. Our data demonstrate that in the intestinal epithelium, CXCR3 is expressed at the luminal level, is overexpressed in CD patients, and co-localizes with gliadin, and that this interaction coincides with recruitment of the adapter protein, MyD88, to the receptor. We also demonstrated that binding of gliadin to CXCR3 is crucial for the release of zonulin and subsequent increase in intestinal paracellular permeability, since CXCR3-deficient mice failed to release zonulin and disassemble TJs in response to a gliadin challenge [32]. Using an α-gliadin synthetic peptide library, we identified two α-gliadin 20mers (QVLQQSTYQLLQELCCQHLW and QQQQQQQQQQQQILQQILQQ) that bind to CXCR3 and release zonulin.

Proposed involvement of the innate and adaptive immune system in the gut mucosal events leading to break of gluten tolerance and onset of celiac disease. Undigested gliading fragment binds to the CXCR3 receptor leading to zonulin release, increased paracellular permeability, access to gliadin fragments into the lamina propria, activation of the innate immune system that in tunr causes activation of the adaptive immune system that ultimately leads to the autoimmune insult of the gut mucosa typical of the celiac enteropathy
Interestingly, gliadin fully reproduces the effects of EGF on the actin cytoskeleton [59], effects that are very similar to those that we reported for zonulin [60]. Since gliadin increases zonulin release from both intestinal cells [53, 55] and whole intestinal tissues through CXCR3 binding [32], it is likely that the gliadin-related EGF effects are indeed secondary to its capability to induce zonulin release. The involvement of the paracellular pathway for gluten trafficking in the lamina propria has also been corroborated by genetic studies identifying an association of some tight junctional genes with CD [61, 62].
There is solid evidence that gluten can also cross the intestinal barrier through the transcellular pathway once tolerance to gluten has been breached [63]. The transferrin receptor CD71, normally expressed on the basolateral pole of enterocytes, is overexpressed on the luminal pole of the intestinal epithelium in CD patients during active disease, leading to apical-to-basal retrotranscytosis of gliadin peptides complexed with secretory IgA (SIgA) [64]. This retrotranscytosis of SIgA–gliadin complexes protects gliadin fragments from lysosomal degradation, facilitating more harmful gliadin peptides to access the intestinal lamina propria, thereby perpetuating intestinal inflammation initiated by the paracellular passage of these peptides (Fig. 2). To further complicate matters, both innate and adaptive immune mechanisms can directly regulate intestinal permeability.
Toll-like receptors (TLRs) are a class of transmembrane pattern recognition receptors (PRRs) that are important for microbial recognition and control of immune responses. TLR2 is one member of the TLR family that recognizes conserved patterns on both Gram-negative and Gram-positive bacteria. TLR2 is expressed on many cell types throughout the intestine, including epithelial cells [65]. Stimulation of TLR2 in vitro increased transepithelial electrical resistance through protein kinase C activation and translocation of ZO-1 to the tight junction complex [66].
Cytokine-mediated modulation of intestinal tight junctions has been the most extensively studied with the focus on tumor necrosis factor (TNF)-α and interferon (IFN)-γ. The effect of TNF-α on the intestinal barrier has been associated in CD [67], IBD [68], and graft-versus-host disease [53].
CD Pathogenesis: The Innate Immune Response
Cytokines such as interleukin (IL)-15 and IFN-α can prime the innate immune response by polarizing dendritic cells and intraepithelial lymphocytes, essential for activating CD [31]. Recent results suggest that specific gliadin peptides may induce EGF and an IL-15-dependent proliferation of enterocytes, structural modifications, vesicular trafficking alterations, signaling and proliferation, and stress/innate immunity activation [69]. Alfa amylase/trypsin inhibitors (ATIs), molecules that confer pest resistance in wheat, also are essential to the innate immune response by engaging the TLR4-MD2 cluster of differentiation (CD) 14 complex with subsequent upregulation of maturation markers and release of proinflammatory cytokines [70]. Along with the breach of epithelial barrier due to gliadin-mediated zonulin release, these mucosal events facilitate the subsequent access of toxic peptides in the lamina propria: Once there, gliadin induces high levels of IL-8, a neutrophil-activating and chemoattractant chemokine [55, 71], and causing the “perfect storm” for CD enteropathy (Fig. 3).
CD Pathogenesis: The Adaptive Immune Response
An aberrant adaptive immune response emerges from a highly specific interplay between selected gluten peptides and major histocompatibility complex (MHC) class II HLA-DQ2/HLA-DQ8 situated on antigen presenting cells (APC) [72]. After the posttranslational deamidation of gluten peptides by tTG2, these peptides become negatively charged and thus more stable within the MHC class II HLA-DQ2/HLA-DQ8 pocket [73]. This cross talk is influenced by the initial imprinting of the innate immune system through IL-15 upregulation that promotes the CD4+ T cell adaptive immune response [74]. Presentation of gluten to CD4+ T cells carried out by dendritic cells as well as macrophages, B cells, and even enterocytes expressing HLA class II, can promote their recirculation in the lamina propria [75]. The contact of CD4+ T cells in the lamina propria with gluten induces their activation and proliferation, with production of proinflammatory cytokines, metalloproteases, and keratinocyte growth factor (KGF) by mesenchymal cells, which induces cryptal hyperplasia and villous blunting secondary to intestinal epithelial cell death induced by IEL [76]. Moreover, overexpression of membrane-bound IL-15 on enterocytes in active CD overexpresses the NK receptors CD94 and NKG2D by CD3+ IEL [77]. CD crypt hyperplasia results as a consequence of continuous tissue damage and the inability of the stem cells to compensate [78].
Clinical Trials Aimed at Controlling Intestinal Permeability in CD
Once the diagnosis of CD is made, a lifelong gluten-free diet (GFD) is the only available treatment for CD, essential for effective disease control and necessary for mucosal healing and the prevention of complications. There are many barriers to following a GFD because gluten is present in many foods besides bread, such as sauces, gravies, coatings for meat and fish, and as an additive for stabilizing, flavoring, and thickening agents. Another issue related to compliance to the GFD is the possibility of cross-contamination that may result from inadequate storage and production in the field (crop rotation, shipment), in factories (products stored or processed in the same plant), at home, and at restaurants (shared utensils and kitchen equipment) [79]. Given the challenges of adhering to such a strict diet, adherence to a GFD can be extremely difficult and challenging in both pediatric and adult patients with CD [80, 81]. As a consequence, there are high expectations for CD drug-based therapy among CD adult patients and parents of children with CD [82, 83].
These considerations definitely justify efforts made in the last decade in pursuing alternative/integrative therapies for CD that incorporate the recent findings that altered gut permeability is implicated in CD pathogenesis [84]. In vivo studies have shown that adding gliadin to the gut luminal compartment increased zonulin release with subsequent increase in gut paracellular permeability, an effect that was reversed by the zonulin inhibitor AT1001 [85]. Similarly, gliadin induced higher permeability via zonulin in in vitro models from biopsies of healthy subjects and from patients with CD consuming a gluten-free diet [37].
Several clinical trials have been conducted with AT1001 (now called larazotide acetate) in CD patients. A clinical study conducted on 20 CD patients consuming a GFD evaluated a 3-day randomized larazotide acetate treatment versus placebo following a 2.5 g oral gluten challenge. In addition to clearly confirming the safety of larazotide acetate with no severe adverse events or dropouts from the trial due to adverse events, a significant 70% increase in intestinal permeability measured as urinary lactulose/mannitol ratio was observed after gluten challenge only in the placebo group, thus confirming the previously described pre-clinical data [86].
Conflicting results on intestinal permeability emerged from a subsequent study of a 14-day treatment with different doses of larazotide acetate [87]. Eighty-six CD patients were evaluated with a gluten challenge when consuming larazotide acetate (0.25, 1, 4, or 8 mg/dose) or a placebo: No difference in intestinal permeability assessed as urinary lactulose/mannitol ratio was found in the larazotide acetate or placebo groups. The authors interpreted these data within the context of a plausible intrinsic variability of urinary lactulose/mannitol ratio assessment in the outpatient setting. Nevertheless, patients receiving larazotide acetate at the dose of 0.25 and 4 mg experienced less symptoms compared to those receiving placebo, possibly suggesting the existence of permeability-driven symptoms in CD. The clinical improvement of 0.25 mg and 4 mg larazotide acetate was outlined by both the Gastrointestinal Symptom Rating Scale (GSRS) and modified celiac disease (CeD)-GSRS scores. No rise of anti-tTG2 antibodies was recorded in both groups, but it is very likely that this relies on the very short duration of the gluten challenge and/or the low gluten dose used.
In another work from the same team, 184 patients who received a daily gluten challenge for 6 weeks were randomized to receive either larazotide acetate (1, 4, or 8 mg) or placebo [88]. The challenge was intended to correspond to an inadvertent ingestion of gluten (2.7 g/day) in patients well controlled on a GFD. Symptoms were assessed during the study by the GSRS; intestinal permeability was evaluated by urinary lactulose/mannitol ratio. Contrary to previous work, the immune response to gluten was also present immediately after the end of the study, on days 49 and 56. Although the primary endpoint (urinary lactulose/mannitol ratio) did not vary throughout the study between the two groups, both anti-tTG2 antibodies and GI complaints were higher in the placebo group at the end of the study. In terms of secondary endpoints, these results provide room to speculate that an aberrant immune response is directly downregulated by larazotide acetate, a finding with clear clinical consequences. While all three doses (1, 4, or 8 mg) of larazotide acetate were capable of blunting the anti-tTG2 response, only the 1-mg dose provided statistically significant protection from symptoms induced by gluten as evaluated by the GSRS scoring system, especially with regard to the specific complaint about diarrhea.
The largest trial (342 adult patients) with larazotide acetate was conducted by the same group in 2015 [89]. The drug was evaluated in CD patients who had persistent symptoms in spite of consuming a GFD for > 12 months. The randomization process split patients in four groups, according to either three different doses of larazotide acetate (0.5, 1, and 2 mg) or to placebo; a gluten challenge was lacking. The study design consisted of a 4-week placebo run-in, a 12-week treatment, and finally a 4-week placebo run-out phase. The primary endpoint was the comparison of the CeD-GSRS severity scores among the four groups; the analysis showed that the 0.5-mg treatment dose significantly decreased gastrointestinal and systemic complaints compared with placebo. The CeD-GSRS changes from baseline in different groups represented secondary endpoints of the study. Clinical amelioration from baseline CeD-GSRS in the 0.5-mg treatment arm was significantly greater than in the placebo group. With regard to the two higher doses of AT1001, no significant clinical response was observed. When considering anti-tTG2 response during the trial, there was no significant difference between larazotide acetate and placebo. This latter study indicated that larazotide acetate could serve as an effective tool integrating the GFD in order to better control symptoms and to improve quality of lives for CD patients.
In conclusion, all of these phase II trials with larazotide acetate have shown that in roughly 500 patients receiving the drug, no safety concerns have been present. Nevertheless, the real utility of the drug in everyday clinical practice remains to be addressed by further studies.
Conclusions
In the last decades, it has become clear that intestinal paracellular permeability is one of the major actors performing on the stage of autoimmunity. The involvement of the paracellular pathway for gluten trafficking in the lamina propria is supported by genetic studies identifying an association of single-nucleotide polymorphism of tight junction genes with CD. Clinical studies of larazotide acetate in CD have started paving the way for controlling symptoms due to poor compliance with the GFD. Whether this molecule could have relevance for other autoimmune conditions, as either prevention or treatment strategy, remains to be established. Improving knowledge about antigen trafficking might unveil the further mechanisms underlying the loss of immune tolerance and systemic autoimmune responses, and offer future treatment strategy hitting autoimmunity at its very onset.
References
Husby S, Koletzko S, Korponay-Szabo IR, et al. European society for pediatric gastroenterology, hepatology, and nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr. 2012;54:136–160.
Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med. 2010;42:530–538.
Lionetti E, Castellaneta S, Francavilla R, et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med. 2014;371:1295–1303.
Fasano A. Celiac disease—how to handle a clinical chameleon. N Engl J Med. 2003;348:2568–2570.
Tapsas D, Hollén E, Stenhammar L, et al. The clinical presentation of coeliac disease in 1030 Swedish children: changing features over the past four decades. Dig Liver Dis. 2016;48:16–22.
Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920.
Händel N, Mothes T, Petroff D, et al. Will the real coeliac disease please stand up? Coeliac disease prevalence in the German LIFE Child Study. J Pediatr Gastroenterol Nutr. 2018;67:494–500.
Singh P, Arora S, Singh A, et al. Prevalence of celiac disease in Asia: a systematic review and meta-analysis. J Gastroenterol Hepatol. 2016;31:1095–1101.
Parra-Medina R, Molano-Gonzalez N, Rojas-Villarraga A, et al. Prevalence of celiac disease in latin america: a systematic review and meta-regression. PLoS ONE. 2015;10:e0124040.
Ege MJ. The hygiene hypothesis in the age of the microbiome. Ann Am Thorac Soc. 2017;14:S348–S353.
De Re V, Magris R, Cannizzaro R. New insights into the pathogenesis of celiac disease. Front Med. 2017;31:137.
Vancamelbeke M, Vermeire S. The intestinal barrier: a fundamental role in health and disease. Expert Rev Gastroenterol Hepatol. 2017;11:821–834.
Okumura R, Takeda K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm Regen. 2018;38:5.
Rodriguez-Pineiro AM, Bergstrom JH, et al. Studies of mucus in mouse stomach, small intestine, and colon. Gastrointestinal mucus proteome reveals Muc2 and Muc5ac accompanied by a set of core proteins. Am J Physiol Gastrointest Liver Physiol. 2013;305:G348–G356.
Moran AP, Gupta A, Joshi L. Sweet-talk: role of host glycosylation in bacterial pathogenesis of the gastrointestinal tract. Gut. 2011;60:1412–1425.
Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124:3–20.
Duffey ME, Hainau B, Ho S, et al. Regulation of epithelial tight junction permeability by cyclic AMP. Nature. 1981;294:451–453.
Itoh M, Nagafuchi A, Yonemura S, et al. The 220-kD protein colocalizing with cadherins in non-epithelial cells is identical to ZO-1, a tight junction-associated protein in epithelial cells: cDNA cloning and immunoelectron microscopy. J Cell Biol. 1993;121:491–502.
Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788.
Furuse M, Fujita K, Hiiragi T, et al. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occluding. J Cell Biol. 1998;141:1539–1550.
Martin-Padura I, Lostaglio S, Schneemann M, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127.
Ikenouchi J, Furuse M, Furuse K, et al. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171:939–945.
Higashi T, Tokuda S, Kitajiri S, et al. Analysis of the ‘angulin’ proteins LSR, ILDR1 and ILDR2–tricellulin recruitment, epithelial barrier function and implication in deafness pathogenesis. J Cell Sci. 2013;126:966–977.
Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455.
Mazzilli MC, Ferrante P, Mariani P, et al. A study of Italian pediatric celiac disease patients confirms that the primary HLA association is to the DQ(alpha 1*0501, beta 1*0201) heterodimer. Hum Immunol. 1992;33:133–139.
Lundin KE, Wijmenga C. Coeliac disease and autoimmune disease-genetic overlap and screening. Nat Rev Gastroenterol Hepatol. 2015;12:507–515.
Dieli-Crimi R, Cenit MC, Nunez C. The genetics of celiac disease: a comprehensive review of clinical implications. J Autoimmun. 2015;64:26–41.
Williamson IA, Arnold JW, Samsa LA, et al. A high-throughput organoid microinjection platform to study gastrointestinal microbiota and luminal physiology. Cell Mol Gastroenterol Hepatol. 2018;6:301–319.
Bach JF. The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol. 2018;18:105–120.
Rao DA. T cells that help B cells in chronically inflamed tissues. Front Immunol. 2018;9:1924.
Kim SM, Mayassi T, Jabri B. Innate immunity: actuating the gears of celiac disease pathogenesis. Best Pract Res Clin Gastroenterol. 2015;29:425–435.
Lammers KM, Lu R, Brownley J, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR32. Gastroenterology. 2008;135:194–204 e193.
Sellitto M, Bai G, Serena G, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS ONE. 2012;7:e33387.
Tripathi A, Lammers KM, Goldblum S, et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci USA. 2009;106:16799–16804.
Asleh R, Marsh S, Shilkrut M, et al. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ Res. 2003;92:1193–1200.
Wicher KB, Fries E. Prohaptoglobin is proteolytically cleaved in the endoplasmic reticulum by the complement C1r-like protein. Proc Natl Acad Sci. 2004;101:14390–14395.
Drago S, El Asmar R, Di Pierro M, et al. Gliadin, zonulin and gut permeability: effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419.
Kurosky A, Barnett DR, Lee TH, et al. Covalent structure of human haptoglobin: a serine protease homolog. Proc Natl Acad Sci USA. 1980;77:3388–3392.
Wicher KB, Fries E. Haptoglobin, a hemoglobin-binding plasma protein, is present in bony fish and mammals but not in frog and chicken. Proc Natl Acad Sci. 2006;103:4168–4173.
Nielsen MJ, Petersen SV, Jacobsen C, et al. A unique loop extension in the serine protease domain of haptoglobin is essential for CD163 recognition of the haptoglobin-hemoglobin complex. J Biol Chem. 2007;282:1072–1079.
Polticelli F, Bocedi A, Minervini G, et al. Human haptoglobin structure and function—a molecular modelling study. FEBS J. 2008;275:5648–5656.
El Asmar R, Panigrahi P, Bamford P, et al. Host-dependent activation of the zonulin system is involved in the impairment of the gut barrier function following bacterial colonization. Gastroenterology. 2002;123:1607–1615.
Thomas KE, Fasano A, Vogel SN. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in Celiac disease. J Immunol. 2006;176:2512–2521.
Scheffler L, Crane A, Heyne H, et al. Widely used commercial ELISA does not detect precursor of haptoglobin 2, but recognizes properdin as a potential second member of the zonulin family. Front Endocrinol. 2018;5:22.
Kouser L, Abdul-Aziz M, Nayak A, et al. Properdin and factor H: opposing players on the alternative complement pathway “see–saw”. Front Immunol. 2013;4:93.
Rittirsch D, Flierl MA, Nadeau BA, et al. Zonulin as prehaptoglobin2 regulates lung permeability and activates the complement system. Am J Physiol. 2013;304:72.
Shirey KA, Lai W, Patel MC, et al. Novel strategies for targeting innate immune responses to influenza. Mucosal Immunol. 2016;9:1173–1182.
Ahout IM, Brand KH, Zomer A, et al. Prospective observational study in two Dutch hospitals to assess the performance of inflammatory plasma markers to determine disease severity of viral respiratory tract infections in children. BMJ Open. 2017;7:e014596.
Ajamian M, Steer D, Rosella G, Gibson PR. Serum zonulin as a marker of intestinal mucosal barrier function: may not be what it seems. PLoS ONE. 2019;14:e0210728.
Hollande F, Blanc EM, Bali JP. HGF regulates tight junctions in new nontumorigenic gastric epithelial cell line. Am J Physiol Gastrointest Liver Physiol. 2001;280:G910–G921.
van der Merwe JQ, Hollenberg MD, MacNaughton WK. EGF receptor transactivation and MAP kinase mediate proteinase-activated receptor-2-induced chloride secretion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008;294:G441–G451.
Cenac N, Chin AC, Garcia-Villar R, et al. PAR2 activation alters colonic paracellular permeability in mice via IFN-gamma-dependent and -independent pathways. J Physiol. 2004;558:913–925.
Brown GR, Lindberg G, Meddings J, et al. Tumor necrosis factor inhibitor ameliorates murine intestinal graft-versus-host disease. Gastroenterology. 1999;116:593–601.
Clemente MG, De Virgiliis S, Kang JS, et al. Early effects of gliadin on enterocyte intracellular signaling involved in intestinal barrier function. Gut. 2003;52:218–223.
Jelinkova L, Tuckova L, Cinova J, et al. Gliadin stimulates human monocytes to production of IL-8 and TNF-alpha through a mechanism involving NF-kappaB. FEBS Lett. 2004;571:81–85.
Silano M, Vincentini O, De Vincenzi M. Toxic, immunostimulatory and antagonist gluten peptides in celiac disease. Curr Med Chem. 2009;16:1489–1498.
Lammers KM, Khandelwal S, Chaudhry F, et al. Identification of a novel immunomodulatory gliadin peptide that causes interleukin-8 release in a chemokine receptor CXCR57-dependent manner only in patients with coeliac disease. Immunology. 2011;132:432–440.
Sapone A, de Magistris L, Pietzak M, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. 2006;55:1443–1449.
Barone MV, Gimigliano A, Castoria G, et al. Growth factor-like activity of gliadin, an alimentary protein: implications for coeliac disease. Gut. 2007;56:480–488.
Wang W, Uzzau S, Goldblum SE, et al. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113:4435–4440.
Monsuur AJ, de Bakker PI, Alizadeh BZ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet. 2005;37:1341–1344.
Wapenaar MC, Monsuur AJ, van Bodegraven AA, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut. 2008;57:463–467.
Schumann M, Richter JF, Wedell I, et al. Mechanisms of epithelial translocation of the alpha(2)-gliadin-33mer in coeliac sprue. Gut. 2008;57:747–754.
Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. 2008;205:143–154.
Cario E, Rosenberg IM, Brandwein SL, et al. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J Immunol. 2000;164:966–972.
Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology. 2004;127:224–238.
Marafini I, Monteleone I, Di Fusco D, et al. TNF-alpha producing innate lymphoid cells (ILCs) are increased in active celiac disease and contribute to promote intestinal atrophy in mice. PLoS ONE. 2015;10:e0126291.
Noth R, Stuber E, Hasler R, et al. Anti-TNF-alpha antibodies improve intestinal barrier function in Crohn’s disease. J Crohns Colitis. 2012;6:464–469.
Barone MV, Troncone R, Auricchio S. Gliadin peptides as triggers of the proliferative and stress/innate immune response of the celiac small intestinal mucosa. Int J Mol Sci. 2014;15:20518–20537.
Reig-Otero Y, Mañes J, Manyes L. Amylase-trypsin inhibitors in wheat and other cereals as potential activators of the effects of nonceliac gluten sensitivity. J Med Food. 2018;21:207–214.
Cinova J, Palova-Jelinkova L, Smythies LE, et al. Gliadin peptides activate blood monocytes from patients with celiac disease. J Clin Immunol. 2007;27:201–209.
Stamnaes J, Sollid LM. Celiac disease: Autoimmunity in response to food antigen. Semin Immunol. 2015;27:343–352.
van de Wal Y, Kooy YMC, van-Veelen P, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161:1585–1588.
Meresse B, Korneychuk N, Malamut G, et al. Interleukin-15, a master piece in the immunological jigsaw of celiac disease. Dig Dis. 2015;33:122–130.
Shibahara T, Wilcox JN, Couse T, et al. Characterization of epithelial chemoattractants for human intestinal intraepithelial lymphocytes. Gastroenterology. 2001;120:60–70.
Salvati VM, Troncone R, Bajaj-Elliott M, et al. Keratinocyte growth factor and coeliac disease. Gut. 2001;49:176–181.
Tang F, Chen Z, Ciszewski C, et al. Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. J Exp Med. 2009;206:707–719.
Senger S, Sapone A, Fiorentino MR, et al. Celiac disease histopathology recapitulates hedgehog downregulation, consistent with wound healing processes activation. PLoS ONE. 2015;10:e0144634.
See JA, Kaukinen K, Makharia GK, et al. Practical insights into gluten-free diets. Nat Rev Gastroenterol Hepatol. 2015;12:580–591.
Valitutti F, Trovato CM, Montuori M, et al. Pediatric celiac disease: follow-up in the spotlight. Adv Nutr. 2017;8:356–361.
Hall NJ, Rubin G, Charnock A. Systematic review: adherence to a gluten-free diet in adult patients with celiac disease. Aliment Pharmacol Ther. 2009;15:315–330.
Branchi F, Tomba C, Ferretti F, et al. Celiac disease and drug-based therapies: inquiry into patients demands. Digestion. 2016;93:160–166.
Norsa L, Tomba C, Agostoni C, et al. Gluten-free diet or alternative therapy: a survey on what parents of celiac children want. Int J Food Sci Nutr. 2015;66:590–594.
Sturgeon C, Fasano A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers. 2016;4:e1251384.
Gopalakrishnan S, Durai M, Kitchens K, et al. Larazotide acetate regulates epithelial tight junctions in vitro and in vivo. Peptides. 2012;35:86–94.
Paterson BM, Lammers KM, Arrieta MC, et al. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–766.
Leffler DA, Kelly CP, Abdallah HZ, et al. A randomized, double-blind study of larazotide acetate to prevent the activation of celiac disease during gluten challenge. Am J Gastroenterol. 2012;107:1554–1562.
Kelly CP, Green PH, Murray JA, et al. Larazotide acetate in patients with coeliac disease undergoing a gluten challenge: a randomised placebo-controlled study. Aliment Pharmacol Ther. 2013;37:252–262.
Leffler D, Kelly C, Green P, et al. Larazotide acetate for persistent symptoms of celiac disease despite a gluten-free diet: a randomized controlled trial. Gastroenterology. 2015;148:1311–1319.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Fasano is co-founder and stock holder of Alba Therapeutics, a company developing treatments complementary to the gluten-free diet by exploiting gut permeability; Dr. Valitutti has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Valitutti, F., Fasano, A. Breaking Down Barriers: How Understanding Celiac Disease Pathogenesis Informed the Development of Novel Treatments. Dig Dis Sci 64, 1748–1758 (2019). https://doi.org/10.1007/s10620-019-05646-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-019-05646-y