
Abstract
MicroRNAs (miRNAs) are highly conserved, small, 18–25 nucleotide, non-coding RNAs that regulate gene expression at the post-transcriptional level. Each miRNA can regulate hundreds of target genes, and vice versa each target gene can be regulated by numerous miRNAs, suggesting a very complex network and explaining how miRNAs play pivotal roles in fine-tuning essentially all biological processes in all cell types in the liver. Here, we summarize the current knowledge on the role of miRNAs in the pathogenesis and diagnosis of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) with an outlook to the broader aspects of metabolic syndrome. Furthermore, we discuss the role of miRNAs as potential biomarkers and therapeutic targets in NAFLD/NASH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are highly conserved, small, 18–25 nucleotide, non-coding RNAs that regulate gene expression at the post-transcriptional level [1]. In most cases, miRNAs bind to the 3′ un-translated region (UTR) of the target mRNA repressing the translation by destabilizing mRNA and/or silencing translation [1]. However, in some instances they can interact with their targets in a non-3′ UTR-dependent manner [2] and cause the up-regulation of their targets [3, 4].
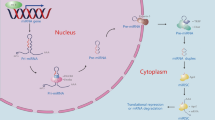
The biogenesis of miRNAs is a strictly controlled, multi-step process [5]. First, miRNAs are transcribed from the genome as pri-miRNAs in the nucleus by RNA polymerase II. Then, pri-miRNAs are cleaved into pre-miRNAs by the DROSHA-DGCR8 complex. The pre-miRNAs are exported to the cytoplasm, where they are cleaved by DICER-TRBP-PACT into mature miRNAs. Finally, the mature guide miRNA strand is loaded onto the RNA-induced silencing complex (RISC) along with AGO2 and GW182 and binds to the target mRNA, while the other strand gets downgraded [5]. However, a Dicer-independent, non-canonical pathway of activation exists too [5].
The first functional miRNA, lin-4, was discovered by Lee et al. [6]. Since then, in the last two decades, the number of miRNAs grew exponentially and today there are thousands of identified mammalian miRNAs [7]. Each miRNA can regulate hundreds of target gene transcripts, and each target gene can be regulated by numerous miRNAs [8]. miRNAs regulate about 50 % of all protein coding genes in mammals [9], fine-tuning essentially all biological processes in all cell types in the liver [8]. Altered hepatic miRNA profile has been described in NAFLD/NASH both in humans and in animal models [10–15].
Here, we summarize the current knowledge on the role of miRNAs in the pathogenesis and diagnosis of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) with an outlook to the broader aspects of metabolic syndrome. Furthermore, we discuss the role of miRNAs as potential biomarkers and therapeutic targets in NAFLD/NASH.
Circulating MicroRNAs as Biomarkers in NASH
The presence of cell-free nucleic acid in plasma and serum has been acknowledged for more than 70 years [16]. While nucleases are found in abundance in the extracellular environment, the association of RNA molecules with proteins, lipids, lipoproteins, etc. increases their stability. AGO2-bound miRNAs can be detected 2 months after cell lysis [17]. The stability of miRNAs in the circulation makes them attractive in biomarker discovery [17–20]. Indeed, several studies have evaluated the specificity of miRNAs and their correlation with liver diseases [16]. miRNAs are found in the circulation in complex with proteins, mainly AGO2, but also AGO1, AGO3, and AGO4 [17, 21] or packaged in extracellular vesicles, mostly exosomes [22, 23]. The release of circulating miRNAs can occur via passive process during cell death or via active release of microvesicles from alive cells [16]. A tissue- and environment-specific process has been suggested by previous studies [24]. We showed that the liver-specific miR-122 is found mostly in the exosome-rich fraction in alcoholic and CpG-induced liver injury, while it is predominant in the protein-rich fraction in acetaminophen-induced toxic liver injury. The inflammatory miR-155 showed similar pattern [25]. This suggests that the distribution of miRNAs in the different compartments, protein versus vesicles, could provide additional information regarding the pathogenesis and increase the likelihood to find a specific miRNA pattern as biomarker. A desirable outcome of biomarker discovery would be to identify miRNAs (or a cluster of them) that could distinguish simple steatosis in NAFLD from steatohepatitis in NASH and/or from NASH with fibrosis.
A recent study assessed circulating miRNAs in NASH patients and found that among 84 circulating miRNAs, miR-122, miR-192, mir-19a, miR-19b, miR-125b, and miR-375 were significantly up-regulated [26]. Comparing simple steatosis with steatohepatitis, the expression of miR-122, miR-192 (up-regulated by TGFβ), and miR-375 (key regulator of glucose homeostasis) correlated with disease severity. Interestingly, the majority of miR-122 was not bound to AGO2 despite that in the lipid loaded hepatocytes miR-122 and AGO2 were co-localized. AGO2 expression differed between patients with simple steatosis and NASH. The authors also found that serum miR-122 and miR-192 positively correlated with serum CK-18 levels and miR-122 performed better than CK18 in predicting liver fibrosis [26].
The increased serum mir-122 levels in NAFLD patients were confirmed by other studies [27, 28] and positive correlation was reported between serum miR-122 levels and hepatic steatosis [27]. In addition higher serum miR-21, miR-34a and miR-451 were found in NAFLD patients [27].
In search of a NAFLD-specific miRNA profile, Tan et al. identified a panel (miR-122 5p, miR-1290, miR-37 3p, miR-192 5p) that showed high diagnostic accuracy and was suggested to be better predictor than ALT or FIB-4 [29].
More importantly, circulating miRNAs are not only “passive” biomarkers. There is increasing evidence that these circulating miRNAs also play important role in the intercellular communication and disease development [30] making them attractive therapeutic targets. Our group recently reported the biodistribution and biological function of extracellular vesicle-associated miR-155 [31].
MiRNA Expression in the Liver in NASH
Although progress has been made in understanding the factors causing NAFLD, more needs to be done to dissect the underlying regulatory networks. Recent evidence indicates the role of miRNAs in energy metabolism [32–37] and liver functions [8]. In addition to the metabolic disturbance, cell death regulation and inflammation are also major determinants of the progression of NAFLD and NASH [38, 39]. Because miRNAs closely regulate cell proliferation and survival as well as the complex process of inflammation, understanding of miRNA circuits and their abnormalities in NASH may promote understanding and modulation of disease.
In human livers with NASH, 23 miRNAs were found to be under- or over-expressed compared to normal livers [10]. The predicted targets of those miRNAs were in cell proliferation, apoptosis, inflammation, oxidative stress, and metabolism. Liver expression of miRNA-122 was significantly decreased in NASH [10]. Notably, differential miRNA expression was found not only in the liver, but also in visceral adipose tissue (VAT) of NAFLD patients [40]. Furthermore, altered gene expression of miRNA processing enzymes (Dicer1, Drosha, DGCR8) was reported in VAT in NASH patients [41].
Numerous animal models are used in NAFLD/NASH research. In livers of ob/ob mice that develop steatosis without prominent inflammation, 8 miRNAs (miR-34a, miR-31, miR-103, miR-107, miR-194, miR-335-5p, miR-221, and miR-200a) were up-regulated and three miRNAs (miR-29c, miR-451, and miR-21) were down-regulated [42]. Together, those findings suggested a connection between miRNAs and metabolic disorders.
High-fat diet (HFD) model that mimics the Western diet, resulting in obesity, insulin resistance, hepatic steatosis and later inflammation also leads to altered miRNA expression. In HFD-fed rats, 44 up-regulated and 12 down-regulated miRNAs were identified [11]. Among those 6 (miR-200a, miR-200b, miR-200c, miR-146a, miR-146b, and miR-152) were up-regulated in vitro in hepatocytes after free fatty acid (FFA) treatment [11]. Studies suggest that the composition of the HFD, including the cholesterol content, affects the miRNA profile [12].
Methionine–choline-deficient (MCD) model of NASH leads to early inflammation, steatohepatitis, and fibrosis, but without the characteristic obesity and peripheral insulin resistance. Our group previously reported a distinct miRNA profile in the livers of MCD diet-fed mice [13]. MCD diet up-regulated 3 % and down-regulated 1 % of the analyzed miRNAs. Five of those miRNAs with altered expression (miR-182, miR-183, miR-199a-3p, miR-705, and miR-1224) were common in alcoholic and nonalcoholic steatohepatitis. All were up-regulated in the MCD model [13]. Recently another group described 71 miRNAs up-regulated (including miRNA-376a, miRNA-127, miRNA-34a, miRNA-300, miRNA-342-3p) and 60 miRNAs down-regulated (including miRNA-122, miRNA-194, miRNA-101b, miRNA-705) in the MCD mouse model [43]. Metformin, which has been shown to improve hepatic steatosis [44], resulted in attenuation of the MCD diet-induced changes in miRNA profile [43]. A recent study from our group found that miR-122 expression was decreased in the liver in the MCD-induced steatohepatitis in mice and the reduction of miR-122 occurred in hepatocytes [28]. Interestingly, expression of pri-miR-122 was also decreased in NASH, suggesting a transcriptional inhibition of miR-122 in NASH. Concomitantly, there was an increase in serum miR-122 levels suggesting loss of liver miR-122 and its potential redistribution to the circulation [28].
Choline-deficient amino acid-define (CDAA) model-induced steatohepatitis and hepatocellular carcinoma (HCC) affects hepatic miRNA expression too. The study of Wang et al. [14] showed differential expression of 30 miRNAs at the different stages of NASH and NASH-related HCC development. The down-regulation of miR-122 and up-regulation of miR-155 (master regulator of inflammation), miR-221/222 [miRNAs involved in epithelial–mesenchymal transition (EMT) and carcinogenesis], and miR-21 (mediator of fibrogenesis) was confirmed by RT-PCR [14]. C/EBPβ and PTEN were identified as potential miR-155 and miR-21 targets, respectively [14].
Alterations of various other mRNAs have been described in human NAFLD/NASH and in animal models of steatohepatitis, and the number is increasing day-by-day.
Functional Effects of miRNA Dysregulation in NASH
While changes in liver miRNA expression in nonalcoholic liver disease and NASH have been reported in human disease as well as in animal models, knowledge is limited regarding the functional significance and causative factors in changes in miRNAs in NASH.
miRNAs in Lipid and Cholesterol Metabolism and Hepatic Steatosis
In the liver, miRNA-122 is expressed in high abundance in hepatocytes where it constitutes for about 80 % of total miRNAs. miR-122 plays multifunctional roles in regulation of lipid metabolism, cell cycle, and in HCV replication [45–48]. It is known to regulate genes involved in fatty acid biosynthesis, and administration of this miRNA antagonist in mice resulted in reduced levels of plasma cholesterol, increased hepatic fatty acid oxidation, and decreased synthesis of hepatic fatty acid and cholesterol [45, 46]. Similarly, miR-122-deficient mice had lower serum cholesterol, LDL, triglyceride, and HDL levels [49, 50]. Silencing of miR-122 in HFD-fed mice reduced hepatic steatosis [45]. However, interestingly, miR-122-deficient mice developed steatohepatitis and HCC despite the lower lipid levels [49, 50]. In humans, miR-122 antagonists are under development.
Hepatic lipid accumulation and inflammation usually occur together. miR-155 is a master regulator of inflammation. Interestingly, mice deficient in miR-155 showed attenuated steatosis but no change in liver damage indicated by serum ALT or inflammation after MCD diet-induced steatohepatitis [51]. Reduction in liver triglycerides and steatosis in miR-155-deficient mice was associated with reduced expression of genes involved in lipid metabolism including Adrp, Dgat2, Cpt1a, Fabp4, Ldtr, Hmgcr, and Ppara [51]. The complexity is well demonstrated by the fact that in another model of NASH, miR-155 deficiency resulted in enhanced hepatic steatosis [52].
miR-21 has been also extensively studied in NASH [53–55]. It has been shown that miR-21 deficiency reduces expression of genes regulating lipogenesis and cell cycle transition via p53 [53]. Furthermore, miRNA-21 inhibition can restore PPARα expression in NASH leading to beneficial outcomes on liver pathology [55].
miR-33a has been found to be involved in fatty acid oxidation, cholesterol homeostasis, and bile acid regulation [56, 57]. The role of miR-33a is complex in these processes as SREBP2 and miR-33a activation resulted in down-regulation of cholesterol hepatic efflux transporters and bile acid synthesis [58]. Furthermore, miR-33-deficient mice develop obesity and liver steatosis presumably due to SREBP1, which is enhanced in the absence of miR-33a [59].
miR-34a has been studied as a potential central factor in NASH [60]. The most prominent target of miR-34a is sirtuin 1 (SIRT1), a NAD-dependent deacetylase that modulates hepatic steatosis and apoptosis [61]. In human NASH, both liver and serum miR-34a were found to be increased and it correlated with the severity of NASH [62].
In HFD-induced steatosis, miR-24 levels were increased in the liver and highly expressed in vitro in hepatocytes; fatty acid treatment up-regulated miR-24 expression [63]. These observations provided basis for additional studies with miR-24-ASO treatment that reduced triglyceride and cholesterol levels in vivo in HFD treated mice [63]; summarized in a recent review of Vincent and Sanyal [64].
miRNAs in Hepatic Inflammation in NASH
There is increasing evidence suggesting the role of innate immunity in NASH [38]. Toll-like receptor (TLR) signaling induces the expression of a variety of miRNAs, and various miRNAs modulate TLR or other PRR signaling both positively and negatively [65].
MiR-155 has been described as a master regulator of inflammation due to its complex effects on key components of intracellular signaling molecules in inflammation [66]. Previous studies from our group have described that miR-155 is increased in Kupffer cells in alcoholic liver disease where it contributes to LPS sensitization and increased TNFα production [3]. Similar to alcoholic liver disease, we found that miR-155 expression was also significantly increased in the livers of mice after MCD diet-induced steatohepatitis [51]. Increased miR-155 was present in hepatocytes as well as in liver mononuclear cells and in Kupffer cells isolated from livers with steatohepatitis compared to control mice. Furthermore, LMNCs and Kupffer cells showed pre-sensitization to further miR-155 increase induced by ex vivo LPS treatment. The pro-inflammatory cytokine, TNFα, is regulated by miR-155 by increasing TNFα mRNA stability and increasing TNFα protein production [3, 66]. Consistent with this, we found that increased miR-155 in LMNC in NASH correlated with increased TNFα production that was further increased by LPS both in LMNC and KCs [51]. In vivo, miR-155 deficiency failed to protect mice from inflammation induced by MCD diet, protein levels of TNFα, MCP-1, and IL-1β, and NF-kB DNA binding was comparable between miR-155 KO and wild-type mice in steatohepatitis [51].
In contrast to miR-155, miR-146a is a negative regulator of TLR signaling and inflammation. While in HFD increased miR-146a expression was reported [11], in the MCD diet model it was significantly down-regulated [67]. Latter one might contribute to the inflammation in NASH via the lack of brake on the pro-inflammatory processes.
Down-regulation of miR-451 was shown to inhibit fatty acid-induced pro-inflammatory cytokine production in NASH via AMPK/Akt pathway [68].
miRNAs in the Progression of NASH and Development of Fibrosis
NASH leads to liver fibrosis and was found to be the etiology of cirrhosis in most patients with “idiopathic” cirrhosis [69]. A broad variety of miRNAs have been identified regulating fibrogenesis via hepatic stellate cell (HSC) activation, EMT etc. The MCD diet model of NASH is characterized by steatosis, inflammation, and development of fibrosis after 6–8 weeks [51] thus widely accepted for studying the progression of NASH.
miR-21 has been extensively studied in fibrosis in NASH [70–72]. It has been shown that miR-21 deficiency reduces expression of genes regulating cell cycle transition via p53 [53]. miR-21 inhibition reduced liver fibrosis by inducing apoptosis of CD24+ progenitor cells [71], and it also regulates ERK1 signaling in HSC activation and hepatocyte EMT [72]. A positive feedback regulation was reported between miR-21 and TGFβ [8].
Fibrosis is the consequence of chronic tissue damage and inflammation, thus we studied the role of miR-155 in NASH fibrosis. Interestingly, miR-155-deficient mice showed reduced fibrosis indicated by attenuated expression of collagen, αSMA and TIMP1, despite the comparable level of inflammation on MCD diet [51]. Reduced fibrosis was associated with reduction in caspase3 protein expression in miR-155 KO mice after MCD diet feeding compared to wild types. The attenuated fibrosis correlated with lower levels of TGFβ and PDGF that both contribute to fibrosis [51]. Further, we found attenuation in liver expression of vimentin and up-regulation of C/EBP DNA binding in miR-155 KO mice compared to wild-type controls with steatohepatitis. Overall, these observations suggested that miR-155 is dysregulated in NASH in multiple cell types in the liver and plays a complex role in regulation of fibrosis in NASH.
miR-34a has been studied as a potential central factor in NASH; this miRNA controls cell cycle arrest, apoptosis, and activation of senescence [61, 73]. The most prominent target of miR-34a is SIRT1, a NAD-dependent deacetylase that modulates apoptosis [61]. Increased miR-34a results in suppression of SIRT1 and subsequent increase in p53 acetylation that leads to increased apoptosis.
While miR-122’s role in lipid metabolism is well established, less data are available on their role in fibrosis. As miR-122-deficient mice develop steatohepatitis, fibrosis, and HCC despite the attenuated dyslipidemia [49, 50], studies investigating the role of miR-122 in fibrosis are warranted. Recently, we identified that reduced miR-122 in the liver and hepatocytes has functional consequences. A miR-122 target, MAP3K3 was increased in livers with steatohepatitis at mRNA level, and this was due to the effect of hepatocyte miR-122 [28] (Fig. 1).

Summarizing our data on the role of miR-122 and miR-155 in NASH
The clinical syndrome of NASH is often associated with obstructive sleep apnea and transient episodes of hypoxia [74, 75], latter one intensively studied in fibrogenesis. We found a significant increase in HIF-1α protein levels as well as in HIF-1α nuclear binding in livers with steatohepatitis and fibrosis after 8 weeks of the MCD diet feeding compared to control mice [28]. Our studies further revealed a causal relationship between HIF-1 increase and miR-122 decreases in NASH. We found that HIF-1α is a miRNA-122 target in hepatocytes, and HIF-1α levels change in a reciprocal direction with miR-122 inhibition or over-expression in hepatocytes [28]. HIF-1α regulates multiple genes at the transcriptional levels. We discovered that parallel to HIF-1 up-regulation in steatohepatitis, the HIF-1α target gene, lysil oxidase, was also significantly increased in livers with steatohepatitis and fibrosis (8-week MCD diet). Liver fibrosis is associated with remodeling. Our study showed that the EMT marker, vimentin was also significantly increased in steatohepatitis. We further showed that vimentin mRNA expression was regulated by miR-122 in hepatocytes from mice with MCD diet-induced steatohepatitis [28]. Together, these results suggested that the decreased hepatic miR-122 levels in MCD diet-induced NASH with fibrosis are associated with increased HIF-1 and vimentin expression and that might contribute to the pathomechanisms of NASH. These new findings highlight the functional role of miR-122 in steatohepatitis that is beyond its originally defined role on regulation of lipid metabolism.
In steatohepatitis induced by alcohol, it was shown that miR-122 is packaged in exosomes and both the numbers of exosomes and miR-122 are increased in the circulation in alcoholic hepatitis in mouse models and in human patients [76]. Furthermore, a functional role for the exosome-packaged miR-122 was identified that indicated that hepatocyte-derived miR-122 is delivered to monocytes and macrophages via exosomes resulting in increases in monocyte/macrophages miR-122 levels. The delivered miR-122 has functional effects on monocytes/macrophages by increasing LPS-induced TNFα production in these inflammatory cells [76]. One can speculate that similar mechanisms may occur in NASH that would provide exosome-mediated intercellular communication between hepatocyte-derived exosomes and liver macrophages via delivery of miR-122 to sensitize the macrophages to inflammatory signals.
Taken together, these observations suggest cell-specific roles for miRNAs in NASH and identify new opportunities for miRNA-based intervention in disease modulation.
MicroRNAs in NASH-Associated HCC
Hepatocellular cancer occurs in livers with cirrhosis where prolonged injury and repair give rise to HCC development. However, in NASH, HCC was found without the presence of cirrhosis both in human disease as well as in animal models suggesting a unique association between NASH and HCC [69]. This association calls for even greater need for biomarkers of HCC. The role of miRNAs in HCC has been the focus of intense investigations both as a mechanistic element in the pathogenesis of HCC and as potential biomarkers [77]. A cluster of up- an down-regulated miRNA have been identified in HCC associated with NASH. Increased levels of serum miR-122 and liver expression of miR-16, miR-33, miR-21, miR-31, miR-221/222, miR-181a/b, let-7a/a, and miR-10b were found [78]. Other miRNAs showed down-regulation in HCC including miR-122 in the liver tissue, miR-34a, miR-200a/b, miR-99am let-7c/g, and miR-199 a/b-3p (summarized by Gori et al.) [78]. Some of the potential targets via miRNAs promote or inhibit hepatocarcinogenesis are the following: the tumor suppressor gene C/EBPβ (miR-155) [14], PTEN (miR-21) [14] and the HBP1-p53-SREBP1c axis (miR-21) [70]. miR-122 targets numerous genes involved in hepatocarcinogenesis, including cyclin G, c-myc, Wnt1, etc. and exhibit an anti-tumor function [79].
Because of these associations, it is not unexpected that miRNA-based therapies are under investigations. In preclinical models, many of these dysregulated miRNAs have been targeted as summarized in by Callegari et al. [80]. Most of the preclinical approaches are based on attempts to restore miR-122 or miR-124. Others aim at inhibition of miR-221 or miR-494; outcomes of these preclinical studies will be informative for further considerations in human disease modification.
MicroRNAs in Obesity and Adipose Tissue Homeostasis
Obesity, the excessive expansion of white adipose tissue, results in chronic low-grade inflammation, and the adipocytes along with adipose tissue macrophages secrete multiple mediators that act in an autocrine/paracrine or endocrine manner. These adipokines are crucial in the cross talk between the adipose tissue and other organs, such as the liver and in the development of insulin resistance and metabolic syndrome [32, 33].
Obesity results in unique or dysregulated miRNA profile, and some of those differentially regulated miRNAs correlate with body weight and some metabolic parameters [32, 33, 81–84]. miRNAs can regulate adipocyte differentiation [85], adipose tissue inflammation [33], as well as hypothalamic regulation of energy homeostasis [35].
miR-132 activates IL-8 and MCP-1 expression via NF-κB in human pre-adipocytes [86]; miR-221 and miR-222 positively correlate with TNFα expression [87], while others have negative effect. miR-126 and miR-193b directly or indirectly inhibit CCL2 secretion, respectively [88]. miR-223 suppresses adipose tissue inflammation via inhibiting infiltration of M1 polarized macrophages [89] and miR-883b-5p via repressing LPS-binding protein (LBP) and TLR4 signaling [90]. Furthermore, miRNAs, beyond regulating adipokine secretion, can be secreted in microvesicles released from adipocytes [91].
miRNAs in Intestinal Homeostasis and Microbiome
There is increasing evidence that changes in gut microbiome are associated with disease development [92], including obesity [93, 94] and NAFLD/NASH [95–98]. The liver’s unique circulation allows direct blood influx from the gastrointestinal tract, and as a first pass organ, it is exposed to the highest concentration of gut-derived microbial components [96]. Increased gut permeability is well-known feature of NAFLD [99, 100]. Inflammasome-associated changes in gut microbiome, including expansion of the Porphyromonadaceae family, results in enhanced steatosis and inflammation via increased influx of TLR4 and TLR9 ligands [95]. Furthermore, deficiency of toll-like receptor 5 (TLR5) affects the composition of gut microbiota and leads to the development of metabolic syndrome [101]. The fact that probiotics might be useful in the therapy of steatohepatitis [102, 103] further supports that gut microbiome has significant influence on the pathogenesis of NASH.
MicroRNAs modulate the expression of genes involved in microbial recognition, PRRs and their downstream signaling, and vice versa, miRNAs are induced by microbial products [104]. The crucial role of miRNAs in the regulation of intestinal homeostasis and gut permeability is well demonstrated by the fact that mice with intestinal epithelial cell (IEC)-specific Dicer deficiency have increased gut permeability [105]. In a pro-inflammatory environment, IECs expressed higher level of miR-122, which increased intestinal permeability via directly targeting occludin [106]. Among others, miR-375 and miR-146a were also implicated in IECs homeostasis [104]. On the role of miRNAs in intestinal immune cells, only few studies are available. miR-155 KO mice have defective intestinal humoral response and delayed bacterial clearance [107]. Our group previously reported that miR-155 deficiency prevented alcohol-induced endotoxin increase in circulation, as well as inflammation in proximal intestine [108]. Similar studies in NASH are warranted. Others have suggested that miR-155 might have anti-inflammatory effect, unique function in intestinal LP T cells [109]. miR-29 in the intestinal dendritic cells targets Th17 response [110], and Th17 axis has been implicated in the pathogenesis of obesity and NAFLD [111]. miR-10a, highly expressed in Treg cells, was suggested to play role in maintenance of immune tolerance [112].
Overall, these suggest that miRNAs might contribute to NASH pathogenesis via regulating the gut microbiome and intestinal homeostasis.
miRNAs in the Metabolic Syndrome
NAFLD/NASH is strongly associated with metabolic syndrome or syndrome X, a cluster of conditions leading to advanced atherosclerosis and cardiovascular diseases as first described by Reaven GM [113]. There are several existing definitions, but all include central obesity, insulin resistance (increased fasting plasma glucose or previously diagnosed type 2 diabetes), dyslipidemia (increased plasma triglycerides and/or low HDL cholesterol), and high blood pressure [114]. Beyond these basic criteria, a link has been implicated between metabolic syndrome and numerous other diseases, such as hyperuricemia and gout [115], asthma [116] and obstructive sleep apnea (OSA) [117], disorders of the reproductive system (polycystic ovary syndrome (PCOS) in females [118], erectile dysfunction in males [119]), psoriasis [120, 121], microalbuminuria and chronic kidney disease [122], certain type of cancers [123], etc.
The role of miRNAs in obesity and dyslipidemia has been discussed above. miRNAs involved in insulin resistance were recently reviewed by others [37]. The role of miRNAs in cardiovascular diseases, being involved in every stage of atherosclerosis, from endothelium dysfunction, cellular adhesion, plaque development, progression, rupture, and thrombus formation, has been recently summarized by Nishiguchi et al. [124]. The role of miRNAs in asthma [125, 126], PCOS [127], psoriasis [128], and chronic kidney diseases [129] has been recently reviewed elsewhere and is beyond the focus of our review.
Here, we briefly discuss the role of miRNAs in hyperuricemia as it has been implicated in the pathogenesis of steatohepatitis. There is increasing evidence that hyperuricemia plays role not only in gout but also in other systemic diseases, including atherosclerosis [124] and alcoholic steatohepatitis [130, 131]. Uric acid crystals can serve as endogenous danger signals and activate the inflammasome complex [132], a pro-inflammatory multiprotein platform that has been previously shown to play role in the pathogenesis of NAFLD/NASH [133]. Uric acid also induces inflammation in adipose tissue via the reduction of adiponectin [134]. Furthermore, uric acid results in insulin resistance and hepatic steatosis by generation of mitochondrial oxidative stress in the liver and direct effect on pancreatic islet cells [135]. Fructose, a major component of the Western diet, increases nucleotide turnover and thus uric acid generation [135]. Elevated serum uric acid levels were found to correlate with hepatic fat content [136, 137].
There are only few studies investigating the role of miRNAs in relation to hyperuricemia. MicroRNA-146a expression was induced by monosodium urate (MSU) crystals in one study, and over-expression of miR-146a resulted in blunted pro-inflammatory cytokine including IL-1β, TNFα, MCP-1, and IL-8 production [19]. Interestingly, while PBMCs from patients with acute gout were shown to have increased miR-146a expression [138], we found significantly lower serum miR-146a levels in NAFLD patients (unpublished observation). Notably, NAFLD is a chronic condition, and in the abovementioned study, miR-146a levels were increased in acute gout, but not in hyperuricemia [138]; furthermore, we do not have data on miR-146a expression in PBMCs in NAFLD. Hyperuricemia was also shown to inhibit angiogenesis [139] and endothelial cell migration [140] via miR-92a and miR-663, respectively. In fatty liver, early activation of angiogenesis positively correlates with fibrosis [141]. Overall these suggest that uric acid might activate pro-inflammatory processes but turn on brakes as well via regulation of certain miRNAs.
Summary
MicroRNAs represent newly discovered regulators and potential biomarkers in the NAFLD and NASH. Due to their pleiotropic functions in regulation of most biological processes, dysregulation of miRNAs contribute to pathogenesis of NAFLD/NASH at various levels of disease development and progression (Fig. 1). Some miRNAs regulate glucose and lipid metabolisms, while others govern cell death and survival pathways. There is a lot more to learn about the role of miRNAs in NASH-associated cell survival and inflammation as they relate to liver fibrosis and remodeling. Progress is on the way in defining potential biomarkers that could aid clinicians to differentiate between benign fatty liver in NAFLD and steatohepatitis with and without hepatocyte necrosis. Further understanding of the meaning and potential role of dysregualted miRNAs, particularly of those in the systemic circulation, will provide better understanding of the overall disease process and aim in development of potential miRNA-based new therapeutic interventions.
Key Points
-
MicroRNAs are small, non-coding RNAs that regulate gene expression at post-transcriptional level.
-
Each mature miRNAs can target many genes, fine-tuning essentially all biological processes in all cell types, including in the liver.
-
Disease-specific tissue miRNA signatures have been identified in various liver diseases including NAFLD, NASH, and HCC.
-
MicroRNAs contribute to pathogenesis of NAFLD/NASH at various levels of disease development and progression.
-
MicroRNA-based therapies are under investigations in preclinical trials.
-
The stability of miRNAs in the circulation makes them attractive in biomarker discovery.
-
A desirable outcome would be to identify miRNAs (or a cluster of them) that could distinguish simple steatosis in NAFLD from steatohepatitis in NASH and/or from NASH with fibrosis.
References
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355.
Bartel DP. MicroRNA target recognition and regulatory functions. Cell. 2009;136:215–233.
Bala S, Marcos M, Kodys K, et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor alpha (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286:1436–1444.
Orom UA, Nielsen FC, Lund AH. Microrna-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–471.
Ohtsuka M, Ling H, Doki Y, Mori M, Calin GA. MicroRNA processing and human cancer. J Clin Med. 2015;4:1651–1667.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854.
Londin E, Loher P, Telonis AG, et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci USA. 2015;112:E1106–E1115.
Szabo G, Bala S. MicroRNAs in liver diseases. Nat Rev Gastroenterol Hepatol. 2013;10:542–552.
Maffioletti E, Tardito D, Genarelli M, Bocchio-Chiavetto L. Micro spies from the brain to the periphery: new clues from studies on microRNAs in neuropsychiatric disorders. Front Cell Neurosci. 2014;8:75.
Cheung O, Puri I, Eicken C, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48:1810–1820.
Feng YY, Xu XQ, Ji CB, Shi CM, Guo XR, Fu JF. Aberrant hepatic microRNA expression in nonalcoholic fatty liver disease. Cell Physiol Biochem. 2014;34:1983–1997.
Karere GM, Glenn JP, VandeBerg JL, Cox LA. Differential microRNA response to a high-cholesterol, high-fat diet in livers of low and high LDL-C baboons. BMC Genom. 2012;13:320.
Dolganiuc A, Petrasek J, Kodys K, et al. MicroRNA expression profile in Lieber–DeCarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol Clin Exp Med. 2009;33:1704–1710.
Wang B, Majumder S, Nuovo G, et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152–1161.
Hoekstra M, van der Sluis RJ, Kuiper J, Van Berkel TJ. Nonalcoholic fatty liver disease is associated with an altered hepatocyte microRNA profile in LDL receptor knockout mice. J Nutr Biochem. 2012;23:622–628.
Enache LS, Enache EL, Ramiere C, et al. Circulating RNA molecules as biomarkers in liver disease. Int J Mol Sci. 2014;15:17644–17666.
Turchinovich A, Weiz L, Langheinz A, Bruwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011;39:7223–7233.
Chen X, Ba Y, Ma L, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006.
Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513–10518.
Weber JA, Baxter DH, Zhang S, et al. The microRNA spectrum in 12 body fluids. Clin Chem. 2010;56:1733–1741.
Arrojo JD, Chevillet JR, Kroh EM, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. 2011;108:5003–5008.
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–433.
Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012;21:R125–R134.
Turchinovich A, Burwinkel B. Distinct AGO1 and AGO2 associated miRNA profiles in human cells and plasma. RNA Biol. 2012;9:1066–1075.
Bala S, Petrasek J, Mundkur S, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology. 2012;56:1946–1957.
Pirola CJ, Fernandez-Gianotti T, Castano GO, et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64:800–812.
Yamada H, Suzuki K, Ichino N, et al. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin Chim Acta. 2013;424:99–103.
Csak T, Bala S, Lippai D, et al. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int.. 2015;35:532–541.
Tan Y, Ge G, Pan T, Wen D, Gan J. A pilot study of serum microRNAs panel as potential biomarkers for diagnosis of nonalcoholic fatty liver disease. PLoS ONE. 2014;9:e105192.
Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659.
Bala S, Csak T, Momen-Heravi F, et al. Biodistribution and function of extracellular miRNA-155 in mice. Sci Rep. 2015;5:10721.
Arner P, Kulyte A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat Rev Endocrinol. 2015;11:276–288.
Ge Q, Brichard S, Yi X, Li Q. microRNA as a new mechanism regulating adipose tissue inflammation in obesity and as a novel therapeutic strategy in the metabolic syndrome. J Immunol Res. 2014; ID: 987285.
Abente EJ, Subramanian M, Ramachandran V, Najafi-Shoushtari SH. MicroRNAs in obesity-associated disorders. Arch Biochem Biophys. 2015. doi:10.1016/j.abb.2015.09.018.
Schneeberger M, Gomez-Valades AG, Ramirez S, Gomis R, Claret M. Hypothalamic miRNAs: emerging roles in energy balance control. Front Neurosci. 2015;9:41.
Chen H, Lan HY, Roukos DH, Cho WC. Application of mircroRNAs in diabetes mellitus. J Endocrinol. 2014;222:R1–R10.
Chakraborty C, Doss CGP, Bandyopadhyay S, Agoramoorthy G. Influence of miRNA in insulin signaling pathway and insulin resistance: micro-molecules with major role in type-2 diabetes. WIREs RNA. 2014;5:697–712.
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846.
Than NN, Newsome PN. A concise review of non-alcoholic fatty liver disease. Atherosclerosis. 2015;239:192–202.
Estep M, Armistead D, Hossain N, et al. Differential expression of miRNAs in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2010;32:487–497.
Sharma H, Estep M, Birerdinc A, et al. Expression of genes for microRNA-processing enzymes is altered in advanced non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2013;28:1410–1415.
Li S, Chen X, Zhang H, et al. Differential expression of microRNAs in mouse liver under aberrant energy metabolic status. J Lipid Res. 2009;50:1756–1765.
Katsura A, Morishita A, Iwama H, et al. MicroRNA profiles following metformin treatment in a mouse model of non-alcoholic steatohepatitis. Int J Mol Med. 2015;35:877–884.
Kita Y, Takamura T, Misu H, et al. Metformin prevents and reverses inflammation in a non-diabetic mouse model of nonalcoholic steatohepatitis. PLoS ONE. 2012;7:e43056.
Esau C, Davis S, Murray SF, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98.
Elmen J, Lindow M, Schutz S, et al. LNA-mediated microRNA silencing in nonhuman primates. Nature. 2008;452:896–899.
Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. miR-122—a key factor and therapeutic target in liver disease. J Hepatol. 2015;2:448–457.
Wilson JA, Sagan SM. Hepatitis C virus and human miR-122: insights from the bench to the clinic. Curr Opin Virol. 2014;7:11–18.
Tsai WC, Hsu SD, Hsu CS, et al. MicroRNA-122 plays critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884–2897.
Hsu SH, Wang B, Kota J, et al. Essential metabolic, antiinflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122:2871–2883.
Csak T, Bala S, Lippai D, et al. MicroRNA-155 deficiency attenuates liver steatosis and fibrosis without reducing inflammation in a mouse model of steatohepatitis. PLoS ONE. 2015;10:e0129251.
Miller AM, Gilchrist DS, Nijjar J, et al. MiR-155 has a protective role in the development of non-alcoholic hepatosteatosis in mice. PLoS ONE. 2013;8:e72324.
Sun C, Huang F, Liu X, et al. miR-21 regulates triglyceride and cholesterol metabolism in non-alcoholic fatty liver disease by targeting HMGCR. Int J Mol Med. 2015;35:847–853.
Ahn J, Lee H, Jung CH, Ha T. Lycopene inhibits hepatic steatosis via microRNA-21-induced downregulation of fatty acid-binding protein 7 in mice fed a high-fat diet. Mol Nutr Food Res. 2012;56:1665–1674.
Loyer X, Paradis V, Henique C, et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARα expression. Gut. 2015. doi:10.1136/gutjnl-2014-308883.
Najafi-Shoushtari SH, Kristo F, Li Y, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;28:1566–1569.
Allen RM, Marquart TJ, Albert CJ, et al. miR-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol Med. 2012;4:882–895.
Li T, Francl JM, Boehme S, Chiang JYL. Regulation of cholesterol and bile acid homeostasis by the CYP7A1/SREBP2/miR-33a axis. Hepatology. 2013;58:1111–1121.
Horie T, Nishino T, Baba O, et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun. 2013;4:2883.
Lee J, Padhye A, Sharma A, et al. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J Biol Chem. 2010;285:12604–12611.
Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–13426.
Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating MicroRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE. 2011;6:e23937.
Ng R, Wu H, Xiao H, et al. Inhibition of microRNA-24 expression in liver prevents hepatic lipid accumulation and hyperlipidemia. Hepatology. 2014;60:554–564.
Vincent R, Sanyal A. Recent advances in understanding of NASH: microRNAs as both biochemical markers and players. Curr Pathibiol Rep. 2014;2:109–115.
Lee HM, Nguyen DT, Lu LF. Progress and challenge of microRNA research in immunity. Front Genet. 2014;5:178.
Tili E, Michaille JJ, Costinean S, Corce CM. MicroRNAs, the immune system and rheumatic disease. Nat Clin Pract Rheumatol. 2008;4:534–541.
Du J, Niu X, Wang Y, et al. MiR-146a-5p suppresses activation and proliferation of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis through directly targeting Wnt1 and Wnt5a. Sci Rep. 2015;5:16163.
Hur W, Lee JH, Kim SW, et al. Downregulation of microRNA-451 in non-alcoholic steatohepatitis inhibits fatty acid-induced proinflammatory cytokine production through the AMPK/Akt pathway. Int J Biochem Cell Biol. 2015;64:265–276.
Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384–1391.
Wu H, Ng R, Chen X, Steer CJ, Song G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut. 2015. doi:10.1136/gutjnl-2014-308430.
Zhang J, Jiao J, Cermelli S, et al. miR-21 inhibition reduces liver fibrosis and prevents tumor development by inducing apoptosis of CD24 + progenitor cells. Cancer Res. 2015;75:1859–1867.
Zhao J, Tang N, Wu K, et al. MiR-21 simultaneously regulates ERK1 signaling in HSC activation and hepatocyte EMT in hepatic fibrosis. PLoS ONE. 2014;9:e108005.
Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–199.
Corey KE, Misdraji J, Gelrud L, et al. Obstructive sleep apnea is associated with nonalcoholic steatohepatitis and advanced live histology. Dig Dis Sci. 2015;60:2523–2528.
Ahmed MH, Byrne CD. Obstructive sleep apnea syndrome and fatty liver: association or casual link? World J Gastroenterol. 2010;16:4243–4252.
Momen-Heravi F, Saha B, Kodys K, Catalano D, Satishchandran A, Szabo G. Increased number of circulating exosomes and their microRNA cargos are potential novel biomarkers in alcoholic hepatitis. J Transl Med. 2015;13:261.
Schutte K, Schultz C, Link A, Malfertheiner P. Current biomarkers for hepatocellular carcinome: surveillance, diagnosis and prediction of prognosis. World J Gastroenterol. 2015;7:139–149.
Gori M, Arciello M, Balsano C. MicroRNAs in nonalcoholic fatty liver disease: novel biomarkers and prognostic tools during the transition from steatosis to hepatocarcinoma. Biomed Res Int. 2014;2014:741465.
Nakao K, Miyaaki H, Ichikawa T. Antitumor function of microRNA-122 against hepatocellular carcinoma. J Gastroenterol. 2014;49:589–593.
Callegari E, Gramantieri L, Domenicali M, D’Abundo L, Sabbioni S, Negrini M. MicroRNAs in liver cancer: a model for investigating pathogenesis and novel therapeutic approaches. Cell Death Differ. 2015;22:46–57.
Chartoumpekis DV, Zaravinos A, Ziros PG, et al. Differential expression of microRNAs in adipose tissue after long-term high fat diet-induced obesity in mice. PLoS ONE 2012;7. ID: e34872.
Ortega FJ, Moreno-Navarrete JM, Pardo G, et al. MiRNA expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS ONE 2010;5. ID: e9022.
Kloting N, Berthold S, Kovacs P, et al. MicroRNA expression in human omental and subcutaneous adipose tissue. PloS ONE 2009;4. ID e4699.
Heneghan HM, Miller N, McAnenea OJ, O’Brian T, Kerin MJ. Differential miRNA expression in omental adipose and in the circulation of obese patients identifies novel metabolic biomarkers. J Clin Endocrin Metab. 2011;96:E846–E850.
Son YH, Ka S, Kim AY, Kim JB. Regulation of adipocyte differentiation via mircoRNAs. Endocrinol Metab (Seoul). 2014;29:122–135.
Strum JC, Johnson JH, Ward J, et al. MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol Endocrinol. 2009;23:1876–1884.
Parra P, Serra E, Palou A, et al. Expression of adipose MicroRNA is sensitive to dietary conjugated linoleic acid treatment in mice. PLoS ONE 2010;5. ID: e13005.
Arner E, Mejhert N, Kulyte A, et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes. 2012;61:1986–1993.
Zhuang G, Meng C, Guo X, et al. A novel regulator of macrophage activation: miR-223 in obesity associated adipose tissue inflammation. Circulation. 2012;125:2892–2903.
Ge Q, Gerard J, Noel L, Scroyen I, Brichard SM. MicroRNAs regulated by adiponectin as novel targets for controlling adipose tissue inflammation. Endocrinology. 2012;153:5285–5296.
Ogawa R, Tanaka C, Sato M, et al. Adipocyte-derived microvesicles contain RNA that is transported into macrophages and might be secreted into blood circulation. BBRC. 2010;398:723–729.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323.
Turnbaugh PJ, Hamadt M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031.
Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185.
Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA. The intestinal microbiota in chronic liver diseases. Adv Immunol. 2013;117:73–97.
Agel B, DiBaise JK. Role of gut microbiome in nonalcoholic fatty liver disease. Nutr Clin Pract. 2015;30:780–786.
Quigley EM, Monsour HP. The gut microbiota and nonalcoholic fatty liver disease. Semin Liver Dis. 2015;35:262–269.
Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772.
Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887.
Vijay-Kumat M, Aitken JD, Carvalho FA, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231.
Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350.
Velayudham A, Dolagniuc A, Ellis M, et al. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology. 2009;49:989–997.
Runtsch MC, Round JL, O’Connell RM. MicroRNAs and the regulation of intestinal homeostasis. Front Genet. 2014;5. ID: 347.
Biton M, Levin A, Slyper M, et al. Epithelial microRNAs regulate gut mucosal immunity via epithelium-T cell crosstalk. Nat Immunol. 1994;12:239–246.
Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:132–1333.
Clare S, John V, Walker AW, et al. Enhanced susceptibility to Citrobacter rodentium infection in mircroRNA-155-deficient mice. Infect Immun. 2013;81:723–732.
Lippai D, Bala S, Catalano D, Kodys K, Szabo G. Micro-RNA-155 deficiency prevents alcohol-induced serum endotoxin increase and small bowel inflammation in mice. Alcohol Clin Exp Res. 2014;38:2217–2224.
Das LM, Torres-Castillo MDLA, Gill T, Levine AD. TGFβ conditions intestinal T cells to express increased levels of miR-155, associated with downregulation of IL-2 and itk mRNA. Mucosal Immunol. 2013;6:167–176.
Brain O, Owens BMJ, Pichulik T, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity. 2013;39:521–536.
Giles DA, Moreno-Fernandez ME, Divanovic S. IL-17 axis driven inflammation in non-alcoholic fatty liver disease progression. Curr Drug Targets. 2015;16:1315–1323.
Takahashi H, Kanno T, Nakayamada S, et al. TGFβ and retinoic acid induce the microRNA miR-10a, which targets bcl-6 and constrains the plasticity of helper T cells. Nat Immunol. 2012;13:587–595.
Reavan GM. Banting lecture 1988: role of insulin resistance in human disease. Diabetes. 1998;37:1595–1607.
Grundy SM, Brewer HB Jr, Cleeman JI, et al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438.
Billiet L, Doaty S, Katz JD, Velasquez MT. Review of hyperuricemia as new marker for metabolic syndrome. ISRN Rheumatol. 2014;2014:852954.
Serafino-Agrusa L, Spatafora M, Scichilone N. Asthma and metabolic syndrome: current knowledge and future perspectives. World J Clin Cases. 2015;3:285–292.
Seetho IW, Wilding JP. Sleep-disordered breathing, type 2 diabetes and the metabolic syndrome. Chron Respir Dis. 2014;11:257–275.
Baldani DP, Skrgatic L, Ouquaq R. Polycystic ovary syndrome: important underrecognized cardiometabolic risk factor in reproductive-age women. Int J Endocrinol. 2015;2015:786362.
Kaya E, Sikka SC, Gur S. A comprehensive review of metabolic syndrome affecting erectile dysfunction. J Sex Med. 2015;12:856–875.
Voiculescu VM, Lupu M, Papagheorghe L, Giurcaneanu C, Micu E. Psoriasis and metabolic syndrome—scientific evidence and therapeutic implications. J Med Life. 2014;7:468–471.
Ganzetti G, Campanati A, Offidani A. Non-alcoholic fatty liver disease and psoriasis: so far, so near. World J Hepatol. 2015;7:315–326.
Prasad GV. Metabolic syndrome and chronic kidney disease: current status and future directions. World J Nephrol. 2014;3:210–219.
Legakis I, Syrigos K. Obesity modulation—the role in carcinogenesis. Anticancer Agents Med Chem. 2010;10:481–490.
Nishiguchi T, Imanishi T, Akasaka T. MicroRNAs and cardiovascular diseases. BioMed Res Int 2015; ID: 682857.
Pua HH, Ansel KM. MicroRNA regulation of allergic inflammation and asthma. Curr Opin Immunol. 2015;36:101–108.
Ebrahimi A, Sadroddiny E. MicroRNAs in lung diseases: recent findings and their pathophysiological implications. Pulm Pharmacol Ther. 2015;34:55–63.
Sorensen AE, Wissing ML, Salo S, Englund AL, Dalgaard LT. MicroRNAs related to polycystic ovary syndrome (PCOS). Genes (Basel). 2014;5:684–708.
Xia J, Zhang W. microRNAs in normal and psoriatic skin. Physiol Genomics. 2014;46:113–122.
Trionfini P, Benigni A, Remuzzi G. MicroRNAs in kidney physiology and disease. Nat Rev Nephrol. 2015;11:23–33.
Iracheta-Vellve A, Petrasek J, Satishchandran A, et al. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J Hepatol. 2015;63:1147–1155.
Petrasek J, Iracheta-Vellve A, Saha B, et al. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells on alcoholic liver disease. J Leukoc Biol. 2015;98:249–256.
Rock KL, Kataoka H, Lai JJ. Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol. 2013;9:13–23.
Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387–400.
Baldwin W, McRahe S, Marek G, et al. Hyperuricaemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes. 2011;60:1258–1269.
Johnson RJ, Nakagawa T, Sanchez-Lozada LG, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–3315.
Sun DQ, Wu SJ, Liu WY, et al. Serum uric acid: a new therapeutic target for nonalcoholic fatty liver disease. Expert Opin Ther Targets. 2015;30:1–13.
Lin H, Li Q, Liu X, et al. Liver fat content is associated with elevated serum uric acid in the Chinese middle-aged and elderly populations: Shanghai Changfeng Study. PLoS ONE. 2015;10:e40379.
Dalbeth N, Pool B, Shaw OM, et al. Role of miR-146a in regulation of the acute inflammatory response to monosodium urate crystals. Ann Rheum Dis. 2015;74:786–790.
Yu S, Hong Q, Wang Y, et al. High concentrations of uric acid inhibit angiogenesis via regulation of the Kruppel-Like-Factor 2-Vascular Endothelial Growth Factor-A axis by miR-92a. Circ J. 2015; PMID: 26299712.
Hong Q, Yu S, Geng X, et al. High concentrations of uric acid inhibit endothelial cell migration via miR-663 which regulates phosphatase and tensin homolog by targeting transforming growth factor-β1. Microcirculation. 2015;22:306–314.
Ciupinska-Kajor M, Hartleb M, Kajor M, et al. Hepatic angiogenesis and fibrosis are common features in morbidly obese patients. Hepatol Int. 2013;7:233–240.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
