
Abstract
Cisplatin resistance is one of the main limitations in the treatment of ovarian cancer, and its mechanism has not been fully understood. The objectives of this study were to determine the role of miR-221/222 and its underlying mechanism in chemoresistance of ovarian cancer. We demonstrated that miR-221/222 expression levels were higher in A2780/CP cells compared with A2780 S cells. An in vitro cell viability assay showed that downregulation of miR-221/222 sensitized A2780/CP cells to cisplatin-induced cytotoxicity. Moreover, we found that knockdown of miR-221/222 by its specific inhibitors promoted the cisplatin-induced apoptosis in A2780/CP cells. Using bioinformatic analysis and luciferase reporter assay, miR-221/222 were found to directly target PTEN. Moreover, knockdown of miR-221/222 in A2780/CP cells significantly upregulated PTEN and downregulated PI3KCA and p-Akt expression. In conclusion, our results demonstrated that miR-221/222 induced cisplatin resistance by targeting PTEN mediated PI3K/Akt pathway in A2780/CP cells, suggesting that miR-221/222/PTEN/PI3K/Akt may be a promising prognostic and therapeutic target to overcome cisplatin resistance and treat ovarian cancer in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epithelial ovarian cancer is the tenth most common cancer and the leading cause of gynecologic cancer-associated deaths among women (Atlanta 2013). Platinum-based chemotherapy is the gold standard for treatment of ovarian cancer, but the high incidence of chemoresistance is considered the greatest barrier to successful treatment (Zheng et al. 2016). Therefore, it is highly important to identify molecular mechanisms to overcome drug resistance.
MiRNAs are a group of small non-coding RNA molecules regulating many protein coding genes by post-transcriptional mechanisms in different cells (Ling et al. 2013). The important roles of miRNAs in regulating various biological processes such as development timing, proliferation, morphogenesis, and apoptosis have been studied in model organisms (Harapan and Andalas 2015). Recent findings have suggested that aberrant miRNA expression promotes chemoresistance and may play a vital role in modulating molecular pathways of drug resistance in cancer cells (Magee et al. 2015). MiR-221 and miR-222 are oncogenic or oncosuppressor miRNAs in human cancers. Up-regulation of these miRNAs has been documented in many types of cancers (Garofalo et al. 2012). More recently, it has been shown that miR-221 and miR-222 are associated with TRAIL-resistant non-small cell lung cancer cells (Garofalo et al. 2009). Also, over expression of miR-221/miR-222 is associated with tamoxifen resistance in breast cancer cells (Miller et al. 2008). Although miR-221/222 are up-regulated in ovarian cancer cells, the role of these miRNAs has not yet been well understood in cisplatin resistance in ovarian cancer. Through targeting several genes that play a part in drug transport pathway, apoptosis, and cell cycle control, miR-221/222 lead to acquisition of drug resistance in different human cancers. Among these genes, phosphatase and tensin homolog (PTEN) expression level is affected dramatically (Li et al. 2016). PTEN is a tumor-suppressor gene, which is located at chromosome 10q23.3 and regulates many key cell processes including growth, adhesion, migration, invasion and apoptosis (Hafsi et al. 2012). MiR-221/222 prevent translation of PTEN through binding to 3′UTR of PTEN mRNA and activates PI3K/AKT pathway. Activation of this pathway leads to inhibition of apoptosis and acquisition of drug resistance in different cancers including ovarian, gastric, and bladder (Cai et al. 2014; Matsuoka and Yashiro 2014; Yuge et al. 2015). Therefore, knockdown or inhibition of miR-221/222 with synthetic oligonucleotides can improve the effects of chemotherapeutic agents on human tumor cells.
In the present study, we investigated whether miR-221/222/PTEN can be used as a novel therapeutic target to overcome cisplatin resistance in ovarian cancer, and then whether inhibition of PI3K/AKT pathway by knockdown of miR-221/222 resensitizes cisplatin-resistant cells to cisplatin.
Materials and methods
Cell culture
Human epithelial ovarian cancer cell lines, A2780 S and A2780/CP, were purchased from the Pasteur Institute of Iran (NCBI, C461 and C454). The cells were grown in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Gibco), 100 U/ml penicillin, and 100 mg/ml streptomycin at 37 °C in a humidified 5% CO2 incubator.
MiRNA inhibitor transfection
A2780/CP cells seeded in 6-well plates were allowed to grow to 70–80% confluency within 2–3 days. The cells were transfected with 100 nM FAM- labeled miR-221/222 inhibitors or scrambled (control) (Life Technologies, Carlsbad, CA, USA) using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. After 24 h, the media were replaced with RPMI-1640 containing 10% FBS. Two days after transfection, the cells were collected for further analysis.
cDNA synthesis and real-time PCR
Total RNA was extracted from the cultured cells using the TRIzol reagent (Invitrogen). Quality of extracted total RNA was evaluated according to 260/280 absorbance ratio, measured by Nano Drop spectrometer (Thermo Scientific, Waltham, MA, USA). Total RNA was converted to cDNA by PrimeScript™ RT Reagent Kit for PTEN mRNA (Takara, Kusatsu, Shiga, Japan). The run method program was set as 37 °C for 30 min, 85 °C for 5 s, and 4 °C for 10 min. Reverse transcription of the miR-221/222 and U6 snRNA (as internal control of miRNA) was performed on total RNA using the “universal cDNA synthesis kit” (Exiqon, Vedbæk, Denmark) in poly A tailing protocol, according to manufacturer protocol. RT-qPCR reactions were performed using the Rotor-Gene 6000 instrument (Corbett Life Science, Australia) according to standard protocols. Briefly, in total volume of 10 μl, 100 ng/μl of miRNA cDNA products was added to a master mix comprising 10 pmol/μl of each miR-221, miR-222, or U6 snRNA DNA primers (Exiqon) and 5U of SYBR premix ExTaq II (TaKaRa). The run method program was set as 95 °C for 10 min followed by 30 cycles at 95 °C for 15 s, 58 °C for 20 s and 72 °C for 25 s. Real-time PCR for PTEN and GAPDH (as endogenous control of mRNA) was performed for 1 cycle at 95 °C for 10 min, followed by 30 cycles at 95 °C for 15 s, 56 °C for 30 s and 72 °C for 30 s. The final concentration of all reagents in the reaction was as follows: 5 × PrimeScript™ Buffer 2 µl, PrimeScript™ RT Enzyme Mix I 0.5 µl, Oligo dT Primer (50 µM) 0.5 µl, Random hexamers (100 µM) 0.5 µl, and RNase Free dH2O 4.5–5.5 µl in total volume of 10 μl. Approximately, 20 ng/μl of cDNA products was added at a later stage. All reactions were performed at least in triplicate. The PCR primers (PTEN and GAPDH) were designed by using Oligo 7 software. The sequences of primers were as follows: PTEN, F: 5′-ACACGACGGGAAGACAAGTT-3′, R: 5′-CTGGTCCTGGTATGAAGAATG-3′; GAPDH, F: 5′-ACGGATTTGGTCGTATTGGG-3′, R: 5′-TGATTTTGGAGGGATCTCGC-3′.
Cell viability assay
Cell viability was determined by MTT assay. A2780/CP cells (8 × 103 cells/well) were transfected with miR-221/222 inhibitors or scrambled oligonucleotide and incubated overnight to allow cell attachment. The blank (not transfected) cells were used as control. Then, cisplatin (0.3–19 µM) was added and incubated at 37 °C. After 48 h, MTT solution (5 mg/ml) was added into each corresponding test well, and the plates were incubated for another 4 h. The culture medium was discarded and DMSO was added. ELISA reader was used to measure the absorbance at 570-nm wavelength. The percentage of cell viability was calculated as Mean of A570 (treated cells)/Mean of A570 (untreated cells) x 100 (Abbosh et al. 2006). All experiments were conducted in triplicate and the results were expressed as mean ± standard deviation (SD).
Apoptosis assay
The apoptosis rate was assessed by the annexin V-FITC apoptosis detection kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s instructions. 6 × 105 A2780/CP cells were seeded in 6-well plates and transfected. After 48 h, the cells were harvested, centrifuged, and washed twice with PBS and double-stained with annexin V-FITC and propidium iodide at room temperature for 15 min and analyzed using flow cytometer (BD Biosciences). After 24 h, the cells were treated with cisplatin at final concentration of 10 µM. The percentages of apoptotic cells were calculated and compared between the transfected and control cells.
Target prediction
Bioinformatic analysis was performed by these software programs: miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/) and Target scan (http://www.targetscan.org/).
Luciferase reporter assay
MiRNA targets were predicted using bioinformatic analysis. The results indicated that miR-221 and miR-222 can bind the same sequence in 3′UTR of PTEN (position 180–206). Therefore, the 3′UTR of the human PTEN gene was PCR amplified with the following primers: PTEN-3′UTR-F, 5′-CGATTCTAGAAATCATGTTCTGGTGG-3′ and PTEN-3′UTR-R, 5′-GCATTCTAGAATTCTGCACAGTAAGCATA-3′ and then cloned into the region directly downstream of the luciferase gene present in the pGL3 vector, to develop the vector PGL3-PTEN. For luciferase assay, A2780/CP cells at the density of 8 × 103 per well in 96-well plates were transfected with 0.2 µg of the PGL3-PTEN or PGL3-control plasmid and 5 pM of miR-221 and/or miR-222 inhibitor using lipofectamine 2000. Cells were harvested 48 h post-transfection and luciferase activity was analyzed using the luciferase assay system (Promega, Madison, WI, USA).
Western blot analysis
Total proteins were extracted from A2780/CP-transfected cells using cold lysis RIPA buffer. All cell lysates were separated by SDS-PAGE (10–12%) and transferred onto PVDF membrane. After being blocked with 5% non-fat dry milk, the membrane was probed with appropriate primary antibody at the dilution of 1:10,000 for PTEN, 1:5000 for p-AKT, and 1:5000 for PI3KCA (Abcam, Cambridge, MA, USA) overnight and secondary antibody (1:5000 dilution) for 1 h. Chemiluminescence system was used to detect the bands. β-actin was used as control.
Statistical analysis
All statistical analysis were performed by GraphPad Prism statistical software, version 5.01 (GraphPad, La Jolla, CA, USA). Each experiment was performed at least in triplicate. All data were expressed as mean ± standard deviation (SD). Real-time PCR data were analyzed using the ΔΔCT method and final data were normalized by u6 or GAPDH expression level as an endogenous controls. One-way ANOVA and Duncan’s test were used to conduct comparisons. p < 0.05 was considered the statistical level of significance.
Results
Altered expression levels of miR-221/222 in cisplatin resistant ovarian cancer cell line, A2780/CP
In this study, we first examined A2780 cell responses to cisplatin using MTT assay. We found that IC50 of cisplatin was 5.1 µM in A2780 S cells and 10 µM in A2780/CP cells. The resistance of A2780/CP cells to cisplatin was 2 times higher than that of the parental cells (Fig. 1a, **p < 0.01). Quantitative RT-PCR was used to assess miR-221, miR-222 and PTEN expression levels in A2780 S and its cisplatin-resistant cell line, A2780/CP. The results showed that miR-221/222 were significantly up-regulated in A2780/CP cells compared with A2780 S cells. The expression level of PTEN in A2780/CP cells was significantly lower than that of A2780 S cells. (Fig. 1b, *p < 0.05, ***p < 0.001).

Expression of miR-221/222 and PTEN in A2780 cells. a Growth inhibition rates of cisplatin for A2780 S and A2780/CP as determined by an MTT assay. Cisplatin treatment decreased ovarian cancer cells growth significantly in a dose-dependent manner. The comparisons of IC50 indicated that A2780 S cells were more sensitive to cisplatin (IC50—5.1 µM) than A2780/CP cells (IC50—10 µM) (**p < 0.01). b RT-PCR results showed that A2780/CP cells had higher expression of miR-221/222 compared with A2780 S cells with the mean increase of 3.5 and 3.25 folds, respectively, compared with A2780 S cells. U6 was used as loading control. Expression of PTEN significantly decreased in A2780/CP cells compared to control cells. GAPDH was used as loading control. Data were expressed as mean and the SD from three independent experiments. (*p < 0.05, ***p < 0.001) (ANOVA)
Blocking miR-221/222 enhance cisplatin-sensitivity in A2780/CP cells
Previous studies reported that miR-221/222 affect sensitivity to cisplatin in vitro and in vivo. To investigate the contribution of miR-221/222 to cisplatin-sensitivity in ovarian cancer cells, A2780/CP cells were transfected with miR-221/222 inhibitors or scrambled oligonucleotide (as a control), respectively, and treated with cisplatin. The transfection efficiency was assessed by fluorescence microscope and flow cytometry. qRT-PCR was used to investigate the miR-221/222 expression levels in A2780/CP cells. The flow cytometric results showed that the transfection efficiency of A2780/CP cells after 24 and 48 h was 22 and 72.22%, respectively (Fig. 2a). According to fluorescence microscope observations, the transfection efficiency after 48 h increased dramatically (Fig. 2b). The qRT-PCR indicated that the miR-221/222 expression levels were significantly lower in miR-221 or miR-222 inhibitor-transfected cells than in the control cells (p < 0.05, Fig. 2c), indicating effective transfection of miR-221/222 inhibitors.

Efficient transfection of miR-221/222 inhibitors in A2780/CP cells a A2780/CP cells were transfected with miR-221 inhibitor, miR-222 inhibitor or scrambled oligonucleotide. A fluorescence microscope and flow cytometry analysis were used to detect the transfection efficiency. As shown in Figs. a and b, a majority of the cells were transfected after 48 h. c Relative expression of miR-221/222 was evaluated by RT-PCR. Expression of miR-221/222 was decreased in A2780/CP-transfected cells versus control cells. The expression of miR-221/222 was at the lowest level 48 h after transfection, indicating effective transfection of miR-221/222 inhibitors (***p < 0.001, ****p < 0.001) (ANOVA)
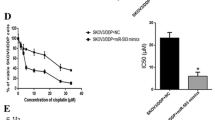
The growth inhibition rate of A2780/CP cells with silencing of miR-221 and/or miR-222 was determined by MTT. Compared with the negative control, miR-221/222 downregulation markedly increased the sensitivity to cisplatin in A2780/CP cells in a dose-dependent manner. IC50 was evaluated as 3.93 μM for miR-221 downregulated group, 3.63 μM for miR-222 downregulated group, and 10 μM for the control in A2780/CP cells. Thus, the IC50 values of miR-221/222 downregulated cells were approximately 2.5 times lower than that of the control cells. In addition, co-treatment with miR-221 and miR-222 inhibitors had stronger inhibitory effect on cisplatin-induced cell cytotoxicity compared with cells transfected with miR-221 or miR-222 inhibitor alone. After co-treatment, the mean IC50 value of cisplatin was reduced to 2.23 μM for A2780/CP cells, confirming that downregulation of miR-221/222 sensitized A2780/CP cells to cisplatin-induced cytotoxicity (p < 0.001, Fig. 3).

Inhibition of miR-221 and miR-222 increases sensitivity of A2780/CP cells to cisplatin. A2780/CP cells were treated with increasing concentrations of cisplatin after being transfected with miR-221/222 inhibitors. After 48 h, MTT assays were performed to assess chemoresistance. Cisplatin-treated group was considered control. We observed that cell viability significantly decreased after cisplatin treatment in A2780/CP-miR-221/222-inhibitor cells compared with control group. Moreover, downregulation of miR-221 and miR-222 induced more significant reduction of cell viability under various concentrations of cisplatin treatment in A2780/CP cells (p < 0.001, ANOVA)
Downregulation of miR-221/222 promote cisplatin-induced apoptosis of A2780/CP cells. Flow cytometric analysis was performed to evaluate the effects of miR-221/222 on apoptosis. The results showed that downregulation of miR-221/222 increased cell apoptosis in A2780/CP cells (Fig. 4). In addition, the significant increase in the counts of annexin V+/PI− cells were found in miR-221/222-transfected cells compared to scramble-transfected or control cells (p < 0.004). An equal apoptosis rate was derived in scramble-transfected cells and the control cells. Interestingly, apoptosis was much higher in cells co-transfected with miR-221 and miR-222 inhibitors compared with those transfected with miR-221 or miR-222 inhibitor.

Inhibition of miR-221/222 promote cisplatin-induced apoptosis of A2780/CP cells. A2780 cells were transfected with miR-221 and/or miR-222 inhibitor and analyzed 48 h later, and apoptosis was measured by annexin V/PI staining. The results showed that transfection of miR-221 or miR-222 inhibitor in A2780/CP cells increased the ratio of apoptotic cells significantly. Interestingly, a strong increase of apoptotic rate was detected in cells transfected with miR-221 and miR-222 inhibitors (p < 0.0001, ANOVA)
MiR-221/222 directly targets PTEN
We performed bioinformatics analysis to predict gene targets of miR-221/222 using miRTarBase and target scan. According to miRTarBase database, 520 and 480 mRNAs were specified as predicted targets of miR-221 and miR-222, respectively. Moreover, all validated targets recovered from the miRTarBase database were approved by convincible experimental evidence such as immunohistochemistry, reporter assay, western blot, and quantitative real time PCR. Our analysis revealed that PTEN was a potential target of miR-221/222 with three complementary sites existing between the 3′UTR of PTEN and miR-221/222 (Fig. 5a). Expression profile of selected miR-221/222 targets was investigate in Unigene database. This analyse showed that PTEN is expressed in ovarian cells. Predicted miR-221/222-target interaction network is shown in Fig. 5b. Accordingly, PTEN is a putative target of miR-221/222. To further investigate whether PTEN was a direct target of miR-221/222, we downregulated miR-221/222 in A2780/CP cells and detected PTEN protein levels by western blot assays. As shown in Fig. 5c, PTEN expression levels increased in miR-221 or miR-222- inhibitor transfected A2780/CP cells compared to control cells. Interestingly, downregulation of miR-221 and miR-222 at the same time, considerably increased the expression level of PTEN in A2780/CP cells in comparison with other groups, suggesting that PTEN can be negatively regulated by miR-221/222, and miR-221/222 modulate cisplatin resistance in human ovarian cancer cells by targeting PTEN.

PTEN is a direct target of miR-221/222 in A2780/CP cells. a The prediction of the binding between miR-221/222 and PTEN by miRTarBase. b Predicted miR-221/222-target interaction network according to strong evidence. c Western blot analysis showed that miR-221/222 inhibition led to significant increase of PTEN in A2780/CP cells. Co-transfection of miR-221 and miR-222 inhibitors into A2780/CP cells considerably upregulated PTEN and downregulated PIK3CA and p-AKT expression compared to other groups. d The luciferase activity of A2780/CP cells was detected after the co-transfection of pGL3-control vector or pGL3-PTEN 3′UTR vector with miR-221 and/or miR-222 inhibitor. There were significant differences in luciferase activity among the groups. Data represent the mean and the SD from three independent experiments (**p < 0.01, ***p < 0.001). (ANOVA)
PTEN is an important negative regulator of the PI3K/Akt pathway, which dephosphorylates PIP3 to make PIP2 and inactivates Akt. To confirm whether miR-221/222 affect the PI3K/Akt signaling pathway in A2780/CP cells, the expression of PI3KCA (the catalytic subunit of PI3K) and phosphorylated Akt (Ser473) were analyzed. Western blot analyses confirmed that levels of PIK3CA and p-AKT proteins were up-regulated in A2780/CP cells, while down-regulated by inhibiting miR-221/222. Additionally, more reduced expression of p-Akt and PIK3CA was reviewed in miR-221 and miR-222-inhibitors treated cells compared to miR-221 or miR-222 inhibitor treated cells.
Next, to validate whether PTEN suppression by miR-221/222 is mediated by direct interaction of miR-221/222 with PTEN-3′UTR, luciferase reporter assay was performed. To this end, the 3′UTR region of PTEN was fused to downstream of the coding sequences for a luciferase reporter in pGL3 vector. Then, A2780/CP cells transiently transfected with this construct in the presence of miR-221 and/or miR-222 inhibitor. As reported in Fig. 5d, co-transfection with miR-221 or miR-222 inhibitor and PGL3-PTEN vector caused an approximately 41.2 and 46.7% increase in luciferase activity compared with PGL3 vector transfected cells (negative control), respectively. Interestingly, when A2780/CP cells were co-transfected with both miR inhibitors and PGL3-PTEN vector, the luciferase activity was significantly increased (p < 0.001) compared with other groups (negative control, PGL3-PTEN/miR-221inhibitor group and PGL3-PTEN/miR-222 inhibitor group), suggesting that miR-221/222 could directly target PTEN 3′UTR. Taken together, these results confirm that miR-221/222 targets PTEN/PI3K/Akt in A2780/CP cells.
Discussion
Chemoresistance is one of the major obstacles to effective ovarian cancer treatment, and efforts to improve the curative effect of conventional chemotherapeutic agents have provided limited results (Kigawa 2013). Recent evidence demonstrates that aberrant miRNAs expression by targeting chemosensitivity or chemoresistance-related genes causes the development of resistance to chemotherapy in various cancers (Chatterjee et al. 2014; Li et al. 2014). Among these, miR-221/222 were identified as two of the most important miRNAs involved in tumor proliferation, invasion and chemoresistance (Garofalo et al. 2012).
Previous studies indicated that miR-221/222 could function as oncogene or oncosuppressor gene in various cancer types. Zhang et al. (2010) reported that miR-221/222 caused decrease in apoptosis in glioblastoma cancer through targeting PUMA. Increased expression of miR-221/222 causes decrease in p27 expression, destruction of cell cycle and increase in cell proliferation in breast cancer and hepatocellular carcinoma (Fu et al. 2011; Miller et al. 2008). Cai et al. (2015) reported that miR-221 upregulation was correlated with poor survival in glioblastomas. However, miR-222 inhibits oral tongue squamous cell carcinoma cell invasion via targeting SOD2 and MMP1 (Liu et al. 2009). Felli et al. showed that miR-221/222 inhibited erythroleukemic cell growth and normal erythropoiesis at least partly via Kit receptor down-modulation (Felli et al. 2005).
In this study, we showed that miR-221/222 were significantly up-regulated in A2780/CP cells. MiR-221/222 over-expression increased the proliferation of cancer cells in vitro. These results suggested that miR-221/222 were oncogenes in ovarian cancer. Recently, miR-221/222 over-expression is related to altered response to conventional chemotherapy. Miller et al.’s study demonstrated that miR-221/222 cause increase in cell proliferation and tamoxifen resistance in basal-type breast cancer through inhibiting cell-cycle inhibitory proteins, i.e. P57 and P27 (Miller et al. 2008). In addition, miR-221/222 cause acquisition of aggressive breast cancer phenotypes with chemoresistance through targeting TRPS1 and increasing epithelial-mesenchymal transition (Stinson et al. 2011). Furthermore, increased expression of miR-221/222 has been reported in TRAIL-resistant cells (Garofalo et al. 2009). MiR-221/222 have been also found to cause induction of fulvestrant resistance in MCF-7 cells through activating β-catenin and inhibiting TGF-β-signaling-induced growth repression (Rao et al. 2011). On the other hand, downregulation of miR-221/222 enhances sensitivity of breast cancer cells to tamoxifen through upregulation of TIMP3 (Garofalo et al. 2012). Liu et al. (2009) indicated that miR-222 inhibitor induced sensitivity to the antitumor effect of sorafenib in human HepG2 cells, confirming that miR-221/222 contribute to drug resistance in various cancers. In consistent with these findings, our results demonstrated that miR-221/222 inhibitors transfection decreased the A2780/CP cell viability, enhanced cisplatin-mediated cytotoxicity, induced apoptosis, and sensitized ovarian cancer cells to cisplatin. Moreover, PTEN was identified as a direct target of miR-221/222 using bioinformatic analysis, and its expression markedly decreased in A2780/CP cells. Our results demonstrated that inhibition of miR-221/222 expression could increase PTEN. Because chemotherapeutic agents are used to induce apoptosis in tumor cells, defective apoptosis pathway can result in development of resistance to apoptosis and failure of chemotherapy. Several genes and regulators play role in development of resistance to apoptosis in cancer cells. Currently, miRNAs’ effects on resistance to apoptosis and, by extension, development of chemoresistance are attracting much attention (Magee et al. 2015). Especially, growing evidence indicates that miR-221/222 cause chemotherapeutic agent-induced apoptosis to be inhibited by targeting the genes that are involved in the pathway of cell cycle control and apoptosis. PTEN is a well-known inhibitor of PI3K/AKT signaling pathway. This pathway is involved in development and progression of tumor and its inappropriate activity causes apoptosis inhibition and chemoresistance development (Hafsi et al. 2012). In the present study, we provided further evidence that miR-221/222 down-regulation led to increase in apoptosis and enhances sensitivity of ovarian cancer cells to cisplatin in A2780 cells through PTEN downregulation and activation of PI3K/AKT pathway.
Consistent with our results, Komeili-Movahhed et al. (2015) confirmed that PI3K/Akt inhibition re-sensitize MCF7 breast cancer cell line to mitoxantrone chemotherapy. Suppression of Rad51 expression or the inactivation AKT signaling overcomes drug resistance to gemcitabine in Human NSCL cells (Tsai et al. 2010). Wang et al. (2016) demonstrated that miR-222 induces adriamycin resistance through PTEN/AKT/p27kip1 pathway in breast cancer. Increased expression of miR-221 leads to increase in cell survival rate, decrease in apoptosis rate, and resistance to cisplatin through PTEN/Akt in osteosarcoma (Zhao et al. 2013). Nagata et al. (2004) found that PTEN-deficient protein could induce the breast cancer trastuzumab resistance through activating PI3K/Akt. Deregulation of the PI3K/PTEN/Akt pathway was associated with resistance to doxorubicin and 4-hydroxyl tamoxifen in breast cancer (Mccubrey et al. 2006). Besides that, any defect of this pathway is involved in drug resistance in prostate cancer (Liu et al. 2015). Activation of PI3K/AKT pathway causes inhibition of cisplatin-induced apoptosis and development of drug resistance in ovarian cancer cells (Lee et al. 2005).
Taken together, our results are consistent with the expected outcomes of an activated PI3K/AKT signaling pathway, and confirm that miR-221/222 contribute to cisplatin resistance in A2780 cells by targeting this pathway.
Conclusion
We demonstrated that knockdown of miR-221/222 could sensitize ovarian cancer cells to cisplatin through upregulating the expression of PTEN that suppresses PI3K/AKT pathway in cancer cells. Our findings suggest that miR-221/222/PTEN/PI3K/AKT may be a promising prognostic and therapeutic target to increase chemosensitivity to cisplatin and treat ovarian cancer.
References
Abbosh PH, Montgomery JS, Starkey JA, Novotny M, Zuhowski EG, Egorin MJ, Moseman AP, Golas A, Brannon KM, Balch C, Huang TH, Nephew KP (2006) Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res 66:5582–5591
Atlanta GA (2013) American Cancer Society. Cancer facts and figures 2013. http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2013
Cai Y, Tan X, Liu J, Shen Y, Wu D, Ren M, Huang P, Yu D (2014) Inhibition of PI3K/Akt/mTOR signaling pathway enhances the sensitivity of the SKOV3/DDP ovarian cancer cell line to cisplatin in vitro. Chin J Cancer Res 26:564–572
Cai G, Qiao Sh, Chen K (2015) Suppression of miR-221 inhibits glioma cells proliferation and invasion via targeting SEMA3B. Biol Res 48:37
Chatterjee A, Chattopadhyay D, Chakrabarti G (2014) MiR-17-5p downregulation contributes to paclitaxel resistance of lung cancer cells through altering beclin1 expression. PLoS ONE 9:e95716
Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F, Liuzzi F, Lulli V, Morsilli O, Santoro S, Valtieri M, Calin GA, Liu CG, Sorrentino A, Croce CM, Peschle C (2005) MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci USA 102:18081–18086
Fu X, Wang Q, Chen J, Huang X, Chen X, Cao L, Tan H, Li W, Zhang L, Bi J, Su Q, Chen L (2011) Clinical significance of miR-221 and its inverse correlation with p27Kip1 in hepatocellular carcinoma. Mol Biol Rep 38:3029–3035
Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A, Taccioli C, Pichiorri F, Alder H, Secchiero P, Gasparini P, Gonelli A, Costinean S, Acunzo M, Condorelli G, Croce CM (2009) miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 16:498–509
Garofalo M, Quintavalle C, Romano G, Croce CM, Condorelli G (2012) miR221/222 in cancer: their role in tumor progression and response to therapy. Curr Mol Med 12:27–33
Hafsi S, Pezzino M, Candido S, Ligresti G, Spandidos DA, Soua Z, McCubrey JA, Travali S, Libra M (2012) Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance. Int J Oncol 40:639–644
Harapan H, Andalas M (2015) The role of microRNAs in the proliferation, differentiation, invasion, and apoptosis of trophoblasts during the occurrence of preeclampsiad. Tzu Chi Med J 27:54–64
Kigawa J (2013) New strategy for overcoming resistance to chemotherapy of ovarian cancer. Yonago Acta Med 56:43–50
Komeili-Movahhed T, Fouladdel S, Barzegar E, Atashpour S, Hossein Ghahremani M, Nasser Ostad S, Madjd Z, Azizi E (2015) PI3K/Akt inhibition and down-regulation of BCRP re-sensitize MCF7 breast cancer cell line to mitoxantrone chemotherapy. Iran J Basic Med Sci 18:472–477
Lee S, Choi EJ, Jin C, Kim DH (2005) Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol 97:26–34
Li B, Ren S, Li X, Wang Y, Garfield D, Zhou S, Chen X, Su C, Chen M, Kuang P, Gao G, He Y, Fan L, Fei K, Zhou C, Schmit-Bindert G (2014) MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 83:146–153
Li B, Lu Y, Wang H, Han X, Mao J, Li J, Yu L, Wang B, Fan S, Yu X, Song B (2016) miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed Pharmacother 79:93–101
Ling H, Fabbri M, Calin GA (2013) MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov 12:847–865
Liu X, Yu J, Jiang L, Shi F, Ye H, Zhou X (2009) MicroRNA-222 regulates cell invasion by targeting matrix metalloproteinase 1 (MMP1) and manganese superoxide dismutase 2 (SOD2) in tongue squamous cell carcinoma cell lines. Cancer Genom Proteom 6:131–139
Liu Z, Zhu G, Getzenberg RH, Veltri RW (2015) The upregulation of PI3K/Akt and MAP kinase pathways is associated with resistance of microtubule-targeting drugs in prostate cancer. J Cell Biochem 116:1341–1349
Magee P, Shi L, Garofalo M (2015) Role of microRNAs in chemoresistance. Ann Transl Med 3:332
Matsuoka T, Yashiro M (2014) The role of PI3K/Akt/mTOR signaling in gastric carcinoma. Cancers (Basel) 6:1441–1463
Mccubrey JA, Steelmana LS, Abramsa SL, Leea JT, Changa F, Bertranda FE (2006) Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Enzyme Regul 46:249–279
Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, Jacob S, Majumder S (2008) MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem 283:29897–29903
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Canc Cell 6:117–127
Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, Burow ME, Ivan M, Croce CM, Nephew KP (2011) MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 30:1082–1097
Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O’Brien C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, Newman RJ, Yue P, Bourgon R, Modrusan Z, Stern HM, Warming S, de Sauvage FJ, Amler L, Yeh RF, Dornan D (2011) TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci Signal 4:ra41
Tsai MS, Kuo YH, Chiu YF, Su YC, Lin YW (2010) Down-regulation of Rad51 expression overcomes drug resistance to gemcitabine in human Non–Small-Cell Lung cancer cells. JPET 335:830–840
Wang DD, Yang SJ, Chen X, Shen HY, Luo LJ, Zhang XH, Zhong SL, Zhao JH, Tang JH (2016) miR-222 induces adriamycin resistance in breast cancer through PTEN/Akt/p27kip1 pathway. Tumour Biol 37:15315–15324
Yuge K, Kikuchi E, Hagiwara M, Yasumizu Y, Tanaka N, Kosaka T, Miyajima A, Oya M (2015) Nicotine induces tumor growth and chemoresistance through activation of the PI3K/Akt/mTOR pathway in bladder cancer. Mol Canc Ther 14:2112–2120
Zhang C, Zhang J, Zhang A, Wang Y, Han L, You Y, Pu P, Kang C (2010) PUMA is a novel target of miR-221/222 in human epithelial cancers. Int J Oncol 37:1621–1626
Zhao G, Cai Ch, Yang T, Qiu X, Liao B, Li W, Ji Z, Zhao J, Zhao H, Guo M, Ma Q, Xiao C, Fan Q, Ma B (2013) MicroRNA-221 induces cell survival and cisplatin resistance through PI3K/Akt pathway in human osteosarcoma. PLoS ONE 8:e53906
Zheng ZG, Xu H, Suo SS, Xu XL, Ni MW, Gu LH, Chen W, Wang LY, Zhao Y, Tian B, Hua YJ (2016) The essential role of H19 contributing to cisplatin resistance by regulating glutathione metabolism in high-grade serous ovarian cancer. Sci Rep 19:26093
Acknowledgements
This work was supported by Shahrekord University of Medical Sciences, Shahrekord, Iran (Research Grant: 1837). We would also like to acknowledge the staffs at the Cellular and Molecular Research Center for their sincere cooperation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interests.
Rights and permissions
About this article

Cite this article
Amini-Farsani, Z., Sangtarash, M.H., Shamsara, M. et al. MiR-221/222 promote chemoresistance to cisplatin in ovarian cancer cells by targeting PTEN/PI3K/AKT signaling pathway. Cytotechnology 70, 203–213 (2018). https://doi.org/10.1007/s10616-017-0134-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-017-0134-z