
Abstract
Current developments in tissue engineering strategies for articular cartilage regeneration focus on the design of supportive three-dimensional scaffolds and their use in combination with cells from different sources. The challenge of translating initial successes in small laboratory animals into the clinics involves pilot studies in large animal models, where safety and efficacy should be investigated during prolonged follow-up periods. Here we present, in a single study, the long-term (up to 1 year) effect of biocompatible porous scaffolds non-seeded and seeded with fresh ex vivo expanded autologous progenitor cells that were derived from three different cell sources [cartilage, fat and bone marrow (BM)] in order to evaluate their advantages as cartilage resurfacing agents. An ovine model of critical size osteochondral focal defect was used and the test items were implanted arthroscopically into the knees. Evidence of regeneration of hyaline quality tissue was observed at 6 and 12 months post-treatment with variable success depending on the cell source. Cartilage and BM-derived mesenchymal stromal cells (MSC), but not those derived from fat, resulted in the best quality of new cartilage, as judged qualitatively by magnetic resonance imaging and macroscopic assessment, and by histological quantitative scores. Given the limitations in sourcing cartilage tissue and the risk of donor site morbidity, BM emerges as a preferential source of MSC for novel cartilage resurfacing therapies of osteochondral defects using copolymeric poly-d,l-lactide-co-glycolide scaffolds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Articular cartilage focal defects represent a common condition affecting the knee and play a significant role in the subsequent development of degenerative joint disease (Huey et al. 2012). The self-healing response to chondral lesions involving the subchondral bone results in fibrocartilage, which has poorer biomechanical and biochemical characteristics compared to native hyaline cartilage (Alford and Cole 2005).
In an attempt to develop tissue engineering strategies, responsive seed cells and supportive scaffolds are sought as they are the critical components for successful resurfacing of articular cartilage. The only approved cell culture-based medicine available today employs autologous articular chondrocytes, which has shown mixed results in humans (Batty et al. 2011) and increases the risk of secondary osteoarthritis due to the procedure followed for tissue harvesting (which requires taking a biopsy of macroscopically intact cartilage from a non-weight bearing part of the joint) (De Bie 2007; Vanlauwe et al. 2011; Zaslav et al. 2009). Recently, much research has invested in searching for alternative cell sources (Oldershaw 2012) including amongst others mesenchymal stromal cells (MSC), which can be readily isolated and expanded in vitro from bone marrow (BM-MSC) or adipose tissue (ASC) (Lee and Im 2010; Pittenger et al. 1999). MSC not only hold high expansion and differentiation capacity but also the potential to recapitulate chondrogenesis when implanted in osteochondral defects (Caplan 1991). Indeed this has been extensively reported in the recent literature [please refer to Veronesi et al. (2014) and Tang et al. (2012) for comprehensive reviews on BM-MSC and ASC in preclinical studies], highlighting the broad use of small animal models in early research stages but also the limited number of studies on large, skeletally mature animal models with prolonged follow-up time periods and undergoing surgical approaches similar to human clinical practice (i.e. arthroscopy) (Hurtig et al. 2011).
In the present work, (A) the feasibility and long-term safety of a cell-based therapy was investigated and (B) the potential of cartilage resurfacing capacity of three different ovine cell sources in combination with poly-d,l-lactide-co-glycolide (PLGA) three-dimensional scaffolds was compared in a large animal model of single-site critical size osteochondral defects.
Materials and methods
Animals
Eight healthy 3-year old ewes of the Ripollesa-Lacona breed were supplied by Servei de Granges i Camps Experimentals (SGCE, Bellaterra, Spain). During the pre- and post-operative procedures, animals were housed together, fed a standard diet and allowed access to water ad libitum. All animal care and experimental procedures were approved by the Universitat Autònoma de Barcelona’s Ethical Committee on Human and Animal Experimentation (Ref. No. CEAAH 501), and registered by the Departament de Medi Ambient de la Generalitat de Catalunya (Reg. No. 3666).
Study design
Two surgical procedures were performed in each animal. The first procedure involved the harvesting of tissues from three different sources (cartilage, BM and fat) for subsequent cell isolation and expansion. The second procedure was performed arthroscopically 4 weeks later and involved the creation of a standardized critical size osteochondral defect bilaterally in the medial and lateral femorotibial condyles. Each of the osteochondral defects produced in the medial and lateral femorotibial condyles were included randomly in either one of any of the three treatment or control groups (Table 1). After surgery, animals were allowed to move freely. Animals were further divided in two new groups, according to the two time end-points (6 and 12 months post-treatment, respectively). Each condyle was considered as an experimental unit, having four experimental units in each animal. A full necropsy was performed on all animals from the 12-month group.
Anaesthesia and post-operative care
All procedures were performed using aseptic techniques and under general anaesthesia. After premedication with an intramuscular (IM) injection of 0.01 mg/kg of buprenorphine (Buprex, Schering-Plough, S.A.) and 0.2 mg/kg of midazolam (Dormicum, Roche), intravenous (IV) access was established at cephalic vein. Animals were pre-oxygenated and induced with 4 mg/kg IV propofol (1 % Propofol-Lipuro, BBraun Melsungen AG). The animals were intubated and maintained on 2 % isoflurane (Isoflo, Abbott laboratories Ltd) mixed with oxygen. Esophageal intubation was made to prevent ruminal bloat. A continuous infusion of Ringer lactate (Ringer lactate, BBraun Melsungen AG) was administered at 10 mL/kg/h during surgery. Intra-operative monitoring consisted of electrocardiography, pulse oximetry, non invasive blood pressure and capnography. All animals received one daily dose of subcutaneous 0.2 mg/kg meloxicam (Metacam, Boehringer) daily for 10 days and a single dose of transdermal 100 µg fentanyl (Durogesic, Janssen-Cilag) for post-operative pain relief. For peri-operative infection prophylaxis, all animals received 22 mg/kg of cefazolin IM (Kurgan, Normon Laboratories) and thereafter every 12 h for 10 days.
Isolation of cells, culture and seeding of scaffolds
Bone marrow was aspirated from the sternum of the animal models and were used for the isolation of the BM-MSC as described elsewhere (Caminal et al. 2014a; Vélez et al. 2012). An articular cartilage layer of 1 mm thick and 6 mm long was harvested from the head of humerus, using a curette. Then cartilage was pierced and used as a source of chondrocytes. Intra-abdominal fat tissue was used as a source of ASC. Tissues were washed and soaked with phosphate-buffered saline solution (PBS, HyClone) and immediately processed in the laboratory. The fragments of fat and cartilage were minced, washed three times in PBS, and digested with 0.2 % (w/v) Collagenase (Sigma-Aldrich) I or II (respectively) for 24 h. Cells and tissues were resuspended in 10 mL of Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10 % (v/v) foetal calf serum (FCS, Biological Industries) and centrifuged at 400 g for 5 min. Cells were washed in PBS and cultured in a T-25 flask in DMEM containing 10 % (v/v) FCS, in a humidified 5 % CO2 incubator. Cells were scaled-up by seeding T-150 flasks at 2,500 cell/cm2. Cell number and viability were determined by the haemocytometer-based trypan blue dye exclusion assay. Glucose and lactate concentrations in supernatants were determined using an YSI 2700 automated analyser (Yellow Springs Instruments). Generation time (td, days), cell growth rate (µ, days−1), specific consumption (qGlc, nmol/106 cells/h) and production rates (qlac, nmol/106 cells/h) of extracellular metabolites (glucose and lactate, respectively) were calculated as described elsewhere (Schop et al. 2009).
ASC, MSC and chondrocyte progenitors were cultured in low glucose DMEM medium supplemented with streptomycin (0.167 g/L, Sigma-Aldrich), kanamycin (0.075 g/L, Sigma-Aldrich) and 10 % FCS. Ex vivo expanded cells were then loaded onto three-dimensional PLGA scaffolds that were manufactured in-house following a solution-casting/salt-leaching technique using PLGA particles with a viscosity of 0.55–0.75 dL/g (Lactel), as described elsewhere (Fonseca et al. 2014). Briefly, polymeric PLGA particles (at 50:50 or 75:25 ratio of PLA:PGA, respectively) were first dissolved in chloroform. Then, sieved salt particles ranging 74–147 µm (for small pores), 300–500 µm (for large pores) and a combination of both (for mixed size pores) were dispersed in the polymer solution at a 9:1 ratio (NaCl:PLGA). The mixture was casted into a cylindrical mould, with a diameter of 4 and 7 mm high. After the evaporation of the solvent, the salt particles were extracted by washing the polymer with distilled water for 48 h. Then the scaffolds were dried at air temperature for 24 h and then vacuum dried for 24 h. Finally, the scaffolds were gamma sterilized (20 kGy) and stored at −20 °C.
For the manufacture of cell:scaffold constructs, 5 × 106 fresh cells (not previously cryopreserved) and scaffolds were incubated for 24 h at 37 °C in stirred minibioreactors (Hexascreen Culture Technologies S.L.) in a final volume of 12 mL of DMEM supplemented with 10 % FCS. Degradation assays were conducted placing the scaffolds in DMEM using a cell culture incubator. Then, samples were taken, lyophilised and weighted, and plotted in order to determine the weight percent of initial polymer.
Differentiation assay
Multipotentiality of isolated cells was determined using StemPro differentiation media (Gibco). Safranin O (Sigma), Oil Red O (Sigma), Alkaline Phosphatase (Takara Bio Inc.) and Von Kossa (Silver nitrate, Sigma) stainings were performed to determine the outcome of the differentiation assays.
Flow cytometry
Flow cytometric analyses were performed as reported elsewhere (Caminal et al. 2014b) in order to evaluate the expression of CD44 (G44-26, BD Biosciences), CD90 (5E10, BD Biosciences), CD105 (MHCD10504, Invitrogen), CD140a (αR1, BD Biosciences), CD166 (3A6, BD Pharmingen) in a FACSCalibur flow cytometer (BD Biosciences). PE-conjugated IgG1 (X40, BD Biosciences) and FITC-conjugated IgG2bk (27-35, BD Biosciences) antibodies were used as isotype controls. Fc-binding receptors were blocked with normal mouse IgG (10400C, Life Technologies).
RT-PCR
Total RNA was extracted using RNeasy Mini columns (Qiagen) according to the manufacturer’s instructions. RNA samples were subjected to reverse transcription-PCR (RT-PCR) analysis in a single-step procedure using the Titan One Tube RT-PCR System (Roche). Reverse transcription was carried out at 50 °C with specific primers, followed by hot-start PCR in the same tube. Primers used were: GAPDH: 5′-CGGATTTGGTCGTATTGG-3′ forward and 5′-TCAAAGGTGGAGGAGTGG-3′ reverse (861 bp); type-I collagen 5′-CCACCAGTCACCTGCGTACA-3′ forward and 5′-GGAGACCACGAGGACCAGAA-3′ reverse (460 bp); type-II collagen 5′-ACGGTGGACGAGGTCTGACT-3′ forward and 5′-GGCCTGTCTCTCCACGTTCA-3′ reverse (141 bp); aggrecan 5′-CCGCTATGACGCCATCTGCT-3′ forward and 5′-TGCACGACGAGGTCCTCACT-3′ reverse (375 bp); biglycan 5′-CCATGCTGAACGATGAGGAA-3′ forward and 5′-CATTATTCTGCAGGTCCAGC-3′ reverse (204 bp); and TGF-β 5′-CGGCAGCTGTACATTGACTT-3′ forward and 5′-AGCGCACGATCATGTTGGAC-3′ reverse (271 bp).
Scanning electron microscopy (SEM)
Samples were fixed in 2.5 % glutaraldehyde and 2 % paraformaldehyde in PBS (pH 7) and dehydrated in ethanol series (15 min in each 30–50–70–90–100–100 %) to absolute ethanol and immediately transferred to acetone before being critical-point dried and gold-coated using a Sputter Coater (K550, Coating Attachment, Emitech Ashford). Samples were examined with an HITACHI S-570 electron microscope at a voltage of 15 kV.
Osteochondral defect creation and arthroscopic implantation
Osteochondral defects were created and test items were implanted arthroscopically. Each joint was approached via a stab incision lateral to the distal aspect of the patellar ligament, allowing the insertion of the arthroscope with its camera for the visualization of the medial condyle. A mechanical shaver was used to remove the fat pad and thus allowing a clearer view. For this purpose, a second stab incision was made medial to the distal aspect of the patellar ligament to allow the shaver insertion. Then the shaver was removed and a mosaicplasty instrument set consisting of a donor and a receptor cannulae (Smith and Nephew Inc), was used to create the defect and to place the scaffold using the cylindrical hollow punch with an inner diameter of 2.7 mm (donor cannula) placed through the second stab incision. This instrument was used to perform a cylindrical osteochondral lesion of diameter 3.5 and 5 mm depth in the caudal aspect of the medial and lateral femoral condyles of each knee. Cylindrical scaffolds were placed through the receptor cannula, filling the lesion by the application of moderate force to the syringe piston. The joints were irrigated by saline solution (NaCl-0.9, BBraun Medical SA) at room temperature during surgery.
Magnetic resonance imaging (MRI)
MRI examination was performed on both knees of each animal under general anaesthesia at 6 or 12 months, according to the experimental group, using a 0.2 T unit with open permanent magnet (Vet-MR, Esaote S.p.a.) as described previously (Fonseca et al. 2014).
Histological evaluation and immunohistochemical procedures
All animals were sedated with an IM injection of 0.02 mg/kg of midazolam and euthanised by IV injection of 12 mg/kg pentobarbital (Dolethal 20 mg/100 mL, Vetoquinol S.A.). Both stifle joints were harvested and condyles fixed in 4 % paraformaldehyde solution at room temperature for 2 weeks, decalcified with 5 % formic acid for 4–6 weeks and embedded in paraffin for subsequent histological and immunohistochemical procedures. Microtome sections 4 μm thick were cut in the sagittal plane and stained with either haematoxylin/eosin (H&E, Sigma-Aldrich) or safranin O (Sigma-Aldrich). The success of the treatment was evaluated blindly following a histological scoring grading scale (Mainil-Varlet et al. 2003) (Suppl. Table 1) on at least 4 sequential histological sections including the areas depicted in Suppl. Figure 1.
The presence of type-II collagen was detected immnunohistochemically using polyclonal antibodies (MAB 8887, Chemicon). Briefly, paraffin sections were deparaffinised, hydrated, and then treated with 0.1 % Pepsin (Sigma-Aldrich) in 0.01 N HCl for 20 min. Endogenous peroxidase activity was blocked with hydrogen peroxide for 30 min and washed in 3 % bovine serum albumin (BSA, Sigma-Aldrich) in PBS. Sections were incubated with the primary antibody (dilution 1:200) overnight at 4 °C. Antibody binding was visualized by using Universal LSAB™2 Kits (Dako) in combination with diaminobenzidine (DAB) according to the manufacturer’s instructions and counterstained with haematoxylin.
Results
Autologous progenitor cells were harvested from ASC, BM and cartilage followed by in vitro expansion (Fig. 1a–c). A total of 600 cm2 (that is four T-150 flasks per cell line) were used in the scale up. The yield ranged 2.1–8 × 104 chondrocyte progenitor cells/cm2, 0.8–5.4 × 104 BM-MSC/cm2, and 0.5–5.2 × 104 ASC/cm2. MSC showed fibroblastic morphology, trilineage potency and expressed CD44, CD90, CD105, CD140a and CD166 surface markers (Fig. 1d, f–h). Expanded cells from cartilage tissue expressed low levels of type-II collagen, as opposed to native articular cartilage used as control, and displayed trilineage potential (Fig. 1e, i–k) Expanded chondrocyte progenitors and ASC displayed higher metabolism than BM-MSC, although similar cell growth rates were observed in the three cell types (Table 2), which were subsequently used for seeding three-dimensional scaffolds. Prior to do this, the degradation profile of different scaffold designs (PLA:PGA ratio and pore size) and cell seeding kinetics were tested with expanded chondrocyte progenitors (Fig. 2). With respect to the degradation profile, no changes in mass were detected within the first 5 weeks. However, at 7 weeks, mass was reduced to half of its initial value in PLGA 50:50 with large pore size (Fig. 2a). Only PLGA scaffolds with large pore sizes (in the range 300–500 µm) were homogeneously colonised with cells, whereas scaffolds with smaller pores displayed cells only on the external surface (Fig. 2b–d). The scaffold used for subsequent studies was PLGA 50:50. In order to confirm the biocompatibility of the scaffolds prior to their use in vivo, these were seeded with expanded chondrocyte progenitors and the metabolic activity of cells loaded onto the PLGA scaffolds was determined by measuring glucose consumption and lactate production for the next 12 days (Fig. 2e). Steady consumption of glucose and production of lactate was observed along the culture time indicative of a viable cell culture. Histological analysis of the scaffolds confirmed the presence of viable cells consistently distributed within the PLGA structure (Fig. 2f). Then, each PLGA scaffold and 5 × 106 cells expanded from either fat, BM or cartilage were incubated in single-use bioreactors resulting in the seeding of an average of 2.28 × 106 ASC (range 1–4.5 × 106 cells), 2.34 × 106 expanded chondrocyte progenitor cells (range 0.7–4 × 106 cells) and 3.37 × 106 BM-MSC (range 1.4–5 × 106 cells), as determined by indirect counting of non-seeded cells present in the supernatant (Table 1).

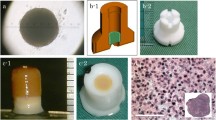
Harvesting of tissues, cell expansion and characterisation. First, tissue was harvested from fat (a), bone marrow (b) and cartilage (c). Then, enzymatic disaggregation was performed for fat and cartilage, and mononuclear cells were isolated from bone marrow and then plated in T-flasks. Finally, ASC, BM-MSC and dedifferentiated chondrocytes were expanded in vitro. Immunophenotype profile of MSC using antibodies that cross-reacted with ovine samples (d) and RT-PCR from cultures of expanded cells from cartilage compared to native cartilage tissue (e). Multipotentiality of MSC and progenitor cells expanded from cartilage tissue as determined in chondrogenesis (f), adipogenesis (g, i, respectively) and osteogenesis (h, j, k) assays. Col II type-II collagen. Scale bars = 200 μm

Characterisation of PLGA scaffolds. In a, degradation profile of PLGA scaffolds at 50:50 and 75:25 PLA:PGA ratios and manufactured with different pore size (small, large and mixed). SEM images of the surface of non-seeded scaffolds and Haematoxylin and Eosin-stained histological analyses of cell-seeded PLGA 50:50 scaffolds manufactured with different pore sizes (b: small pore sizes in the range 74–147 µm and 74.96 ± 0.15 % porosity; c: mixed pore sizes in the range 74–147 and 300–500 µm and 81.73 ± 0.02 % porosity; and d: large pore sizes in the range 300–500 µm and 77.06 ± 0.04 % porosity) after 24-h incubation time. Biocompatibility of PLGA scaffolds loaded with cells expanded from cartilage tissue as determined by glucose consumption and lactate production in culture (e). In f, paraffin section of a cell-seeded PLGA 50:50 scaffold after 12 days in culture. In g, chondrogenesis assay for expanded cells from cartilage (samples 1 and 2) and MSC (sample 3) differentiated for 12 days in vitro either in monolayer or loaded onto PLGA scaffolds. Col I type-I collagen; Col II type-II collagen. Scale bars = 400 μm, except in e = 200 μm
Experimental lesion, test item implantation and clinical outcome
Bilateral arthroscopy was well tolerated and all animals were able to stand on all four limbs and walk immediately after surgery. No clinical complications occurred after the transplantation procedures in any of the animals with the exception of F64 (from the 6-month group), that was found dead at month 4 after treatment. The necropsy performed on this animal revealed pneumonia as a most probable cause of death, which was not related to the treatment (Supplemental Fig. 2).
All the constructs remained firmly adhered at the implantation site and PLGA was fully resorbed at the two end-time points of the study, as revealed by macroscopical observation and confirmed by histological analysis, consistent with the degradation rate observed in vitro (Fig. 2a). A full necropsy was performed on all animals from the 12-month group in order to exclude any potential long-term toxicity that may be caused by the treatment. No infections, fibrinous overgrowth on the joint surface or signs of other potential adverse effects were observed in any of the organs.
Untreated and cell-free controls
Major findings in the untreated control group included the presence of fissures extending to the subchondral bone and MRI analyses revealed signs of bone oedema (Fig. 3a). Spongy bone increased its thickness and invaded the cartilage surface. Wide areas of the articular surface were refilled with fibrocartilage and presented focuses of vascularisation (Fig. 3b). The presence of poteoglycans was restricted to the deepest zone, around the columnar-distributed chondrocytes but not on the surface (Fig. 3c). Type-II collagen staining accumulated in S1 but was fainter in D1 (Fig. 3d; Suppl. Figure 1).

Untreated and cell-free scaffold control groups. MRI image from F58l (a), Haematoxylin and Eosin staining of a section from F52l (b), safranin O staining of a section from animal F52l (c) and immunohistochemical staining for type-II collagen on a tissue section from animal F59l (d). MRI image from F44r (e), macroscopic view of the defect site on condyle F44r (f); safranin O staining (g) and immunohistochemical staining for type-II collagen (h) on sequential sections of condyle F44r. Scale bars = 500 μm
With respect to cell-free scaffold controls, at 6 months post-treatment, the presence of a fibrous matrix was restricted to the deepest zone (Table 3; Fig. 3e–h). Integration in S1 and S2 territories was irregular and subchondral bone presented residual calcified cartilage in D1. At 12 months post-treatment, irregularities were observed on the surface at the defect site, not levelled with the surrounding tissue and presenting deep depressions and fissures. MRI analysis showed wider lesions than the defect modelled initially with a marked irregularity in the subchondral bone.
Scaffolds seeded with chondrocytes expanded from cartilage biopsies
At 6 months post-treatment, proteoglycan and type-II collagen content did not match the expression levels observed in neighbouring normal tissue, and the integration was only partial at the S1/D1 border. In S1 and D1, chondrocytes were grouped in clusters as if migrating to the lesioned area. Cystic empty cavities with broad fibrous focuses were observed within the spongy bone. In some cases, lesions appeared fully repaired at the macroscopic level and displayed shiny smooth white tissue of great similarity to the surrounding intact articular cartilage, whilst others presented deep depressions of the surface with a colour that differed from the native adjacent tissue (Table 3). MRI analyses revealed subchondral bone lesions with sclerotic aspect and small osseous oedemas, as evidenced by STIR hypersignal. At 12 months, S1 appeared uniform and levelled in most cases (Fig. 4b). S1 and S2 were made of hyaline cartilage with columnar chondrocytes in the deep zone of S1 and grouped on the surface areas (Fig. 4c). Subchondral bone in D1 and D2 was similar to normal tissue in S3, evidencing complete bone regeneration (Table 4). Type-II collagen content and safranin O staining extent and intensity was higher in all specimens from the 12-month group compared to the 6-month treatment group.

MRI and histological analyses at 12 months post-treatment with PLGA scaffolds loaded with cells. MRI image of condyle F61l (a); Haematoxylin and Eosin staining (b) and safranin O staining of lateral condyle F61l (c); immunohistochemical staining for type-II collagen on medial condyle F61r (d). MRI image of condyle F59r (e); Haematoxylin and Eosin staining of condyle F52r (f); safranin O (g) and immunohistochemical staining for type-II collagen (h) staining of condyle F59r. MRI image of condyle F61l (i) and safranin O staining of condyles F61l (j) and F52r (k) and immunohistochemical staining for type-II collagen of condyle F52r (l). Scale bars = 500 μm, except f = 1,000 μm and k = 200 µm
Scaffolds seeded with adipose tissue-derived mesenchymal stromal cells
At the 6 month time point, massive surface depressions were observed macroscopically on the defect site. MRI analyses confirmed the presence of sclerotic lesions affecting the subchondral lamina. Better results were obtained at 12 months. Histologically, the surface appeared irregular in some samples with vertical fissures that deepened into the middle region of the articular cartilage, but presenting good integration to the perilesional zones S2 and S3 (Fig. 4g, h). Hyaline-like tissue predominated, although fibrous tissue was also present in some areas at the integration border of the subchondral bone and the surface (Fig. 4g). Subchondral bone in D1 and D2 presented increased thickness with large trabeculae and the tidemark (which is the basophilic line between calcified and uncalcified cartilage) disappeared (Fig. 4h). MRI analyses did not evidence any noteworthy improvement, and it was possible to detect lesions with pseudonodular morphology in some specimens (Fig. 4e).
Scaffolds seeded with bone marrow-derived mesenchymal stromal cells
At 6 months post-treatment, MRI analyses did not reveal extensive lesions, but craters were observed on the caudomedial aspect in line with the results obtained histologically. At the end of 12 post-transplantation months, the defects were completely regenerated with white, shiny, and smooth tissue similar to the intact articular cartilage, although some fissures were also observed. MRI analyses showed the presence of smaller lesions than in other treatment groups, as demonstrated by T1 and T2 hiposignal, and absence of canvas STIR sequences (Fig. 4i). No morphological alterations were observed on the condylar surface, which was compatible with a regenerated lesion. Histologically, the extracellular matrix (ECM) was of hyaline nature in S1, with chondrocytes organised individually and in columns (Fig. 4j). Subchondral bone also showed normal aspect. In some specimens, though, the lesion was found partially levelled but thinner with the presence of fissures on the surface (Fig. 4l). Proteoglycan and type-II collagen contents were poor in S1, with scarce cellularity in relation to adjacent native cartilage (Fig. 4h; Table 4).
Discussion
In the orthopaedic field, special attention is given to novel tissue-engineering techniques for the repair of damage on hip, knee and elbow joints, due to the complexity of treating pathologies that involve two types of tissue (namely, subchondral bone and avascular articular cartilage) (Capito and Spector 2003). Despite of the extensive literature on the use of stem cell therapies at the research stage, only ex vivo expanded chondrocytes have been approved by the regulatory authorities for human use and have thus become the gold standard in cell therapy for cartilage resurfacing of focal injuries (Vanlauwe et al. 2011).
In the present study, scaffolds of PLGA (a copolymer made of PLA and PGA) were used as a vehicle of three different cell types that were tested side-to-side under the same experimental conditions in order to assess cartilage resurfacing capacity at 6 and 12 months post-implantation on critical size osteochondral defects. PLGA was chosen because (A) its proven biocompatibility, (B) the feasibility of three-dimensional molding of the constructs in the shape of the experimental defects, (C) it allowed minimally invasive arthroscopic implantation and (D) its degradation rate, which was compatible with the expected deposition of ECM by the cells loaded on the scaffolds (Villalobos Córdoba et al. 2007; Niederauer et al. 2000; Sittinger et al. 1996; Uematsu et al. 2005). Moreover, the use of porous PLGA scaffolds permitted the retention of the cells at the defect site and promoted homogeneous distribution throughout the graft.
We observed that PLGA scaffolds at a ratio of 50 (PLA):50 (PGA) preserved their integrity up to 5 weeks in vitro, which is compatible with the time taken by the cells to synthesize the new ECM [from 9 to 20 days, according to the work performed by Barry and collaborators (Barry et al. 2001)] that substitutes the scaffolding material (Middleton and Tipton 2000). Furthermore, PLGA-based constructs allow arthroscopic implantation, making this biomaterial even more attractive for clinical use, since the arthroscopic technique reduces morbidity, surgical time, patient recovery and secondary complications typically derived from open surgery that are routinely performed in tissue engineering strategies for cartilage repair (Villalobos Córdoba et al. 2007; Erggelet et al. 2007; Niederauer et al. 2000). Although in some samples the tide mark was faint or disappeared, it is remarkable that none of the specimens showed detachment of neocartilage from subchondral bone, indicating that the integration between the two layers resisted the load bearing forces in all treatment groups and highlighting the suitability of PLGA scaffolds in the treatment of osteochondral defects.
With respect to cell types, the differentiation potential of MSC into chondrocytes and osteoblasts makes these cells very attractive for the simultaneous regeneration of bone/cartilage lesions. Additionally, MSC can be expanded extensively in vitro whilst chondrocytes hold a limited culture growth capacity and display phenotypic instability during the course of their expansion in monolayer culture. Such phenotypic instability (also called “dedifferentiation”) is characterised by a shift of cellular morphology from a rounded to the typical fusiform fibroblastic shape, among other features (Schnabel et al. 2002), as we also observed in the present study (Fig. 1i). Considering the advantages of using BM-MSC and provided that both chondrocytes and BM-MSC gave similar results with respect to the quality of the new tissue, our results may suggest that BM-MSC as the preferred cell source in cell-therapies aiming at osteochondral repair using PLGA scaffolds. On the other hand, we observed that ASC were not as effective as BM-MSC to regenerate osteochondral defects neither at 6 nor at 12 months post-treatment. Different expression profiles in ASC and BM-MSC may explain why the latter differentiate more efficiently into bone and cartilage, whereas ASC differentiate better into adipocytes as reported previously by Liu and collaborators (Liu et al. 2007) thus supporting our observations in the present study.
The safety of the implantation of autologous cell:scaffold constructs was demonstrated by the absence of local or systemic adverse effects during the clinical follow-up and by a full necropsy performed at 12 months post-treatment. With respect to the efficacy, the presence of cartilage of hyaline quality 1 year after treatment with either BM-MSC or chondrocytes is a key point since current surgical approaches typically result in a short-term clinical success but eventually fail due to poor mechanical properties of the mixed matrix that is generated. For example, even though fibrocartilaginous repair tissue from microfracture results in initially enhanced clinical knee-function scores at earlier assessment time points, it later degrades, and scores decline (Mithoefer et al. 2009). This also highlights the importance of using large animal models with anatomy similar to the human knee allowing orthopaedic surgeons to undergo procedures similar to those in human practice and therefore making possible to assess the performance of novel implants in situ using minimally invasive approaches. Due to its size and anatomy, sheep arises as a relevant translational animal model for this type of research and, in fact, it has been previously used in several other studies for the treatment of chondral and osteochondral lesions [as comprehensively reviewed by Ahern and collaborators (Ahern et al. 2009)].
Finally, we conclude that the use of expanded cells from cartilage, BM and ASC in combination with PLGA scaffolds for cell therapy of osteochondral defects is safe even after 1 year post-administration and that cartilage of hyaline quality is observed principally after using cells derived from cartilage and BM. Further work is needed in order to evaluate dose–effect relationships, improvement of integration to adjacent tissue by optimising scaffold design and to explore the mechanisms involved in the regeneration process, which may uncover additional advantages of the use of MSC in contrast to chondrocytes, such as MSC’s anti-inflammatory effects.
References
Ahern BJ, Parvizi J, Boston R, Schaer TP (2009) Preclinical animal models in single site cartilage defect testing: a systematic review. Osteoarthr Cartil 17:705–713. doi:10.1016/j.joca.2008.11.008
Alford JW, Cole BJ (2005) Cartilage restoration, part 1: basic science, historical perspective, patient evaluation, and treatment options. Am J Sports Med 33:295
Barry F, Boynton RE, Liu B, Murphy JM (2001) Chondrogenic differentiation of mesenchymal stem cells from bone marrow: differentiation-dependent gene expression of matrix components. Exp Cell Res 268:189–200. doi:10.1006/excr.2001.5278
Batty L, Dance S, Bajaj S, Cole BJ (2011) Autologous chondrocyte implantation: an overview of technique and outcomes. ANZ J Surg 81:18–25. doi:10.1111/j.1445-2197.2010.05495.x
Caminal M, Fonseca C, Peris D, Moll X, Rabanal RM, Barrachina J, Codina D, García F, Cairó JJ, Gòdia F, Pla A, Vives J (2014a) Use of a chronic model of articular cartilage and meniscal injury for the assessment of long-term effects after autologous mesenchymal stromal cell treatment in sheep. N Biotechnol 31:492–498. doi:10.1016/j.nbt.2014.07.004
Caminal M, Moll X, Codina D, Rabanal RM, Morist A, Barrachina J, Garcia F, Pla A, Vives J (2014b) Transitory improvement of articular cartilage characteristics after implantation of polylactide:polyglycolic acid (PLGA) scaffolds seeded with autologous mesenchymal stromal cells in a sheep model of critical-sized chondral defect. Biotechnol Lett. doi:10.1007/s10529-014-1585-3
Capito RM, Spector M (2003) Scaffold-based articular cartilage repair. IEEE Eng Med Biol Mag 22:42–50
Caplan AI (1991) Mesenchymal stem cells. J Orthop Res 9:641–650. doi:10.1002/jor.1100090504
De Bie C (2007) Genzyme: 15 years of cell and gene therapy research. Regen Med 2:95–97. doi:10.2217/17460751.2.1.95
Erggelet C, Neumann K, Endres M, Haberstroh K, Sittinger M, Kaps C (2007) Regeneration of ovine articular cartilage defects by cell-free polymer-based implants. Biomaterials 28:5570–5580. doi:10.1016/j.biomaterials.2007.09.005
Fonseca C, Caminal M, Peris D, Barrachina J, Fàbregas PJ, Garcia F, Cairó JJ, Gòdia F, Pla A, Vives J (2014) An arthroscopic approach for the treatment of osteochondral focal defects with cell-free and cell-loaded PLGA scaffolds in sheep. Cytotechnology 66:345–354. doi:10.1007/s10616-013-9581-3
Huey DJ, Hu JC, Athanasiou KA (2012) Unlike bone cartilage regeneration remains elusive. Science 338:917–921. doi:10.1126/science.1222454
Hurtig MB, Buschmann MD, Fortier LA, Hoemann CD, Hunziker EB, Jurvelin JS, Mainil-Varlet P, McIlwraith CW, Sah RL, Whiteside RA (2011) Preclinical studies for cartilage repair. Cartilage 2:137–152. doi:10.1177/1947603511401905
Lee JS, Im GI (2010) Influence of chondrocytes on the chondrogenic differentiation of adipose stem cells. Tissue Eng Part A 16:3569–3577. doi:10.1089/ten.TEA.2010.0218
Liu TM, Martina M, Hutmacher DW, Hui JH, Lee EH, Lim B (2007) Identification of common pathways mediating differentiation of bone marrow- and adipose tissue-derived human mesenchymal stem cells into three mesenchymal lineages. Stem Cells 25:750–760. doi:10.1634/stemcells.2006-0394
Mainil-Varlet P, Aigner T, Brittberg M, Bullough P, Hollander A, Hunziker E, Kandel R, Nehrer S, Pritzker K, Roberts S, Stauffer E (2003) Histological assessment of cartilage repair: a report by the Histology Endpoint Committee of the International Cartilage Repair Society (ICRS). J Bone Joint Surg Am 85(Suppl 2):45–57
Middleton JC, Tipton AJ (2000) Synthetic biodegradable polymers as orthopedic devices. Biomaterials 21:2335–2346
Mithoefer K, McAdams T, Williams RJ, Kreuz PC, Mandelbaum BR (2009) Clinical efficacy of the microfracture technique for articular cartilage repair in the knee: an evidence-based systematic analysis. Am J Sports Med 37:2053–2063. doi:10.1177/0363546508328414
Niederauer GG, Slivka MA, Leatherbury NC, Korvick DL, Harroff HH, Ehler WC, Dunn CJ, Kieswetter K (2000) Evaluation of multiphase implants for repair of focal osteochondral defects in goats. Biomaterials 21:2561–2574
Oldershaw RA (2012) Cell sources for the regeneration of articular cartilage: the past, the horizon and the future. Int J Exp Pathol 93:389–400. doi:10.1111/j.1365-2613.2012.00837.x
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Schnabel M, Marlovits S, Eckhoff G, Fichtel I, Gotzen L, Vecsei V, Schlegel J (2002) Dedifferentiation-associated changes in morphology and gene expression in primary human articular chondrocytes in cell culture. Osteoarthr Cartil 10:62–70. doi:10.1053/joca.2001.0482
Schop D, Janssen FW, van Rijn LD, Fernandes H, Bloem RM, de Bruijn JD, van Dijkhuizen-Radersma R (2009) Growth, metabolism, and growth inhibitors of mesenchymal stem cells. Tissue Eng Part A 15:1877–1886. doi:10.1089/ten.tea.2008.0345
Sittinger M, Reitzel D, Dauner M, Hierlemann H, Hammer C, Kastenbauer E, Planck H, Burmester GR, Bujia J (1996) Resorbable polyesters in cartilage engineering: affinity and biocompatibility of polymer fiber structures to chondrocytes. J Biomed Mater Res 33:57–63
Tang QO, Carasco CF, Gamie Z, Korres N, Mantalaris A, Tsiridis E (2012) Preclinical and clinical data for the use of mesenchymal stem cells in articular cartilage tissue engineering. Expert Opin Biol Ther 12:1361–1382. doi:10.1517/14712598.2012.707182
Uematsu K, Hattori K, Ishimoto Y, Yamauchi J, Habata T, Takakura Y, Ohgushi H, Fukuchi T, Sato M (2005) Cartilage regeneration using mesenchymal stem cells and a three-dimensional poly-lactic-glycolic acid (PLGA) scaffold. Biomaterials 26:4273–4279. doi:10.1016/j.biomaterials.2004.10.037
Vanlauwe J, Saris DB, Victor J, Almqvist KF, Bellemans J, Luyten FP (2011) Five-year outcome of characterized chondrocyte implantation versus microfracture for symptomatic cartilage defects of the knee: early treatment matters. Am J Sports Med 39:2566–2574. doi:10.1177/0363546511422220
Vélez R, Hernández-Fernández A, Caminal M, Vives J, Soldado F, Fernández A, Pla A, Aguirre M (2012) Treatment of femoral head osteonecrosis with advanced cell therapy in sheep. Arch Orthop Trauma Surg 132:1611–1618. doi:10.1007/s00402-012-1584-6
Veronesi F, Maglio M, Tschon M, Aldini NN, Fini M (2014) Adipose-derived mesenchymal stem cells for cartilage tissue engineering: State-of-the-art in in vivo studies. J Biomed Mater Res A 102:2448–2466. doi:10.1002/jbm.a.34896
Villalobos Córdoba FE, Velasquillo Martínez C, Martínez López V, Lecona Butrón H, Reyes Marín B, Estrada Villaseñor E, Villegas CH, Solís Arrieta L, Espinosa Morales R, Ponce de León CI (2007) Resultados en la reparación experimental de lesiones osteocondrales en un modelo porcino mediante ingeniería de tejidos. Acta Ortop Mex 21:217–223
Zaslav K, Cole B, Brewster R, DeBerardino T, Farr J, Fowler P, Nissen C (2009) A prospective study of autologous chondrocyte implantation in patients with failed prior treatment for articular cartilage defect of the knee: results of the Study of the Treatment of Articular Repair (STAR) clinical trial. Am J Sports Med 37:42–55. doi:10.1177/0363546508322897
Acknowledgments
The authors would like to acknowledge Anna Garrit and Cristian de la Fuente for technical assistance; and José Luís Ruiz, Ramón Costa and the crew of the “Servei de Granges i Camps Experimentals” of the Universitat Autònoma de Barcelona (Bellaterra, Spain) for their careful assistance to animal management. This work was supported by “Ministerio de Economía y Competitividad” (Grant Number IPT-300000-2010-0017), “Ministerio de Ciencia e Innovación” (Grant Numbers PSE-010000-2007-4//PSE-010000-2008-4, BIO2008-01985), “Programa de suport a grups d’investigació DGR/DIUE” (Grant number 2009SGR1038) and by the European Regional Development Fund (ERDF), within the National Plan for Scientific Research, Development and Innovation 2008-2011.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Caminal and D. Peris have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10616_2015_9842_MOESM2_ESM.jpg
SUPPLEMENTAL Fig. 1. Areas analysed in the histological assessment. “S” corresponds to the superficial zone and “D” for the deep zone. The approximate dimensions are indicated. Superficial cartilage zone: S1, defect site; S2, perilesional area; S3, surrounding native hyaline cartilage. Deep zone: D1, damaged area; D2, adjacent spongy bone. (JPEG 194 kb)
10616_2015_9842_MOESM3_ESM.jpg
SUPPLEMENTAL Fig. 2. Necropsy. The only pathological finding in animal F64 was the presence of purulent abscesses in the lung parenchyma (arrow). (JPEG 1605 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Caminal, M., Peris, D., Fonseca, C. et al. Cartilage resurfacing potential of PLGA scaffolds loaded with autologous cells from cartilage, fat, and bone marrow in an ovine model of osteochondral focal defect. Cytotechnology 68, 907–919 (2016). https://doi.org/10.1007/s10616-015-9842-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-015-9842-4