
Thirteen compounds were isolated from the bulbs of Lilium brownii var. viridulum Baker, including a new chlorophenyl glycoside identified as 5-chloro-6-methoxy-4-methylphenyl 1-O-β-D-glucopyranosyl-(1→6)-α-L-rhamnopyranoside. The structure of this new compound was elucidated using comprehensive spectroscopic analyses, acid hydrolysis, and derivatization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
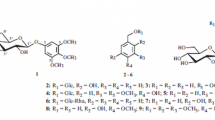
Lilium, an important medicinal herb with a long history, has been widely used in traditional Chinese medicine and the food industry. It can be found in various regions across Asia, Europe, and North America and it is native to China [1]. Lilium possesses multiple medicinal and nutritional values, which include nourishing Yin, moisturizing the lungs, and calming the mind. Previous studies have indicated that Lilium contains at least 82 steroidal saponins, 12 phenylpropanoid glycosides, 5 chlorophenyl glycosides, and some phenyl glycosides [2,3,4]. To further explore the chemical composition and new bioactive compounds from Lilium, we reported a new chlorophenyl glycoside, along with 12 known phenylpropanoid compounds.
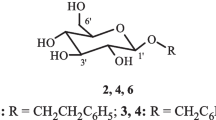
The methanol extract of Lilium bulbs was subjected to both normal and reverse-phase silica column chromatography, as well as preparative HPLC to isolate a new chlorophenyl glycoside (1), 1-O-feruloylglycerol (2) [5], 1-O-p-coumaroylglycerol (3) [6], caffeic acid methyl ester (4) [7], 1-O-p-coumaroyl-3-O-feruloylglycerol (5) [8], 1,3-O-diferuloylglycerol (6) [9], 1-O-feruloy1-3-O-caffeoylglycerol (7) [8], 1-O-caffeoyl-3-O-p-coumaroylglycerol (8) [8], regaloside A (9) [10], regaloside B (10) [10], syringaresinol (11) [11], pinoresinol (12) [11], and epipinoresinol (13) [11]. Compounds 11–13 were isolated from the Lilium genus to our knowledge for the first time.
Compound 1 was obtained as a white amorphous powder. Its molecular formula was deduced as C20H29O11Cl based on the HR-ESI-MS (m/z 479.1337 [M – H]–; calcd 479.1318). Additionally, the isotopic molecular ion peak was observed at m/z 481.1311 with an abundance ratio of 3:1, indicating the presence of one chlorine atom. The 1H NMR spectrum of 1 showed two aromatic protons at δ 6.66 (1H, d, J = 8.0 Hz, H-2) and 6.65 (1H, d, J = 8.0 Hz, H-3), a methoxy proton at δ 3.84 (3H, s, 6-OCH3), a methyl proton at δ 2.33 (3H, s, CH3-4), and two anomeric proton signals at δ 4.86 (1H, d, J = 7.5 Hz, H-1′) and δ 4.70 (1H, d, J = 2.0 Hz, H-1′′). The 13C NMR spectrum of 1 displayed 20 carbon signals, combining with the HSQC and HMBC spectrum, which were assigned as follows: δ 158.0 (C-1), 157.1 (C-6), 139.0 (C-5), 117.1 (C-4), 111.6 (C-3), and 100.9 (C-2) for the six aromatic carbons, 10 oxymethine carbons at δ 102.5 (C-1′), 74.9 (C-2′), 77.9 (C-3′), 71.5 (C-4′), 76.9 (C-5′), 102.1 (C-1′′), 72.1 (C-2′′), 72.4 (C-3′′), 74.1 (C-4′′), and 69.8 (C-5′′), one oxymethylene carbon at δ 67.8 (C-6′), one methoxy carbon at δ 56.7 (6-OCH3), and two methyl carbon at δ 20.7 (CH3-4) and 17.9 (C-6′′). The acid hydrolysis and derivatization of 1, as determined by HPLC analysis, indicated the absolute configuration of the monosaccharide units to be one D-glucose and one L-rhamnose [12]. The coupling constant of the anomeric protons at δ 4.86 (1H, d, J = 7.5 Hz, H-1′) and δ 4.70 (1H, d, J = 2.0 Hz, H-1′′) indicated the presence of a β-configured glucosyl and an α-rhamnosyl [4]. Correlations from H-2 to C-1, C-3, and C-4, from H-3 to C-2, C-4, and CH3-4, from CH3-4 to C-3, C-4, and C-5, and from 6-OCH3 to C-6, as well as correlations from H-1′ to C-1 and from H-6′ to C-1′′ in the HMBC spectrum confirmed the structure of 1 as 5-chloro-6-methoxy-4-methylphenyl 1-O-β-D-glucopyranosyl-(1→6)-α-L-rhamnopyranoside.

Key HMBC and 1H–1H COSY correlations of compound 1.
Experimental
Plant Material. The Lilium were purchased from Jingshiqiao Town, Longhui County, Shaoyang City, Hunan Province in 2020. They were identified as the bulbs of Lilium brownii var. viridulum Baker by Prof. Guo Tao from the School of Pharmacy, Henan University of Chinese Medicine. The voucher specimens are stored at the Henan Engineering Research Center of Medicinal and Edible Chinese Medicine Technology.
Extraction and Isolation. Forty kilograms of fresh Lilium bulbs were extracted twice with 70% ethanol (25 L) for 14 days each time at room temperature. The combined extract was concentrated under reduced pressure and partitioned with petroleum ether, ethyl acetate, and n-butanol. The ethyl acetate fraction (34 g) was obtained. The ethyl acetate fraction was subjected to column chromatography on a silica gel column (700 g, 8 × 77 cm, 200 mesh) using a eluent mixture of dichloromethane–methanol (20:1→2:1). This resulted in the separation of 18 fractions (A1–A18), with each fraction collected under the guidance of a UV detector (210 nm and 254 nm). Fraction A5 was further subjected to reverse-phase chromatography (YMC-pack ODS-A, 10 × 250 mm, S-5 μm) using a mobile phase of MeOH–H2O (60:40, 3 mL/min) to isolate caffeic acid methyl ester (4, 14.3 mg). Fraction A6 was eluted using a mobile phase of MeOH–H2O (46:54, 3 mL/min) to isolate pinoresinol (12, 29.6 mg) and epipinoresinol (13, 3.0 mg). Fraction A7 was eluted using a mobile phase of MeCN–H2O (32:68, 3 mL/min) to isolate 1,3-O-diferuloylglycerol (6, 5.0 mg); using a mobile phase of MeOH–H2O (50:50, 3 mL/min) to isolate syringaresinol (11, 29.6 mg). Fraction A8 was eluted using a mobile phase of MeOH–H2O (40:60, 3 mL/min) to isolate 1-O-feruloylglycerol (2, 127.7 mg). Fraction A9 was eluted using a mobile phase of MeCN–H2O (26:74, 3 mL/min) to isolate 1-O-p-coumaroyl-3-O-feruloylglycerol (5, 5 mg), 1-O-feruloy1-3-O-caffeoylglycerol (7, 10.1 mg), 1-O-caffeoyl-3-O-p-coumaroylglycerol (8, 5.3 mg). Fraction A10 was eluted using a mobile phase of MeOH–H2O (25:75, 3 mL/min) to isolate 1-O-p-coumaroylglycerol (3, 21.6 mg). Fraction A13 was eluted using a mobile phase of MeOH–H2O (34:66, 3 mL/min) to isolate regaloside B (10, 173.0 mg). Fraction A16 was eluted using a mobile phase of MeCN–H2O (18:82, 3 mL/min) to isolate 1 (5.0 mg) and regaloside A (9, 5.3 mg).
Acid Hydrolysis and Derivatization. Compound 1 (1.0 mg) was placed in a solution of 0.5 M HCl (0.2 mL) and heated at 90°C for 2 h. After drying, the reaction mixture was dissolved in anhydrous pyridine (0.2 mL). Then, L-cysteine methyl ester hydrochloride (1 mg) was added. The mixture was heated at 60°C for 1 h. Then, 1 mg of o-tolyl isothiocyanate was added, and the reaction mixture was heated at 60°C for 1 h. After drying the solution, the residue was partitioned between H2O and cyclohexane. The H2O layer was concentrated, dissolved in methanol, and analyzed using semi-preparative HPLC with a mobile phase of 52% methanol. The wavelength was set at 254 nm for detection. The peaks at 15.85 and 20.35 min coincided with derivatives of D-glucose and L-rhamnose in compound 1.
1-O-β-D-Glucopyranosyl-(1→6)-α-L-rhamnopyranoside (1), white amorphous powder, \({\left[\mathrm{\alpha }\right]}_{{\text{D}}}^{20}\) –63.0° (c 0.10, MeOH). UV (MeOH, λmax, nm) (log ε): 210 (2.92). HR-ESI-MS m/z 479.1337 [M – H]– (calcd for C20H28O11Cl, 479.1318). 1H NMR (500 MHz, CD3OD, δ, ppm, J/Hz): 1.21 (3H, d, J = 6.5, H-6′′), 2.33 (3H, s, CH3-4), 3.35–3.37 (1H, m, H-4′), 3.35–3.37 (1H, m, H-4′′), 3.42–3.46 (1H, m, H-3′), 3.49–3.53 (1H, m, H-2′), 3.55–3.60 (1H, m, H-5′), 3.56–3.59 (1H, m, H-6′b), 3.61–3.65 (1H, m, H-5′′), 3.64–3.67 (1H, m, H-3′′), 3.81–3.82 (1H, m, H-2′′), 3.84 (3H, s, OCH3), 4.05 (1H, dd, J = 13.5, 5.0, H-6′a), 4.70 (1H, d, J = 2.0, H-1′′), 4.86 (1H, d, J = 7.5, H-1′), 6.65 (1H, d, J = 8.0, H-3), 6.66 (1H, d, J = 8.0, H-2). 13C NMR (125 MHz, CD3OD, δ, ppm): 17.9 (C-6′′), 20.7 (CH3), 56.7 (OCH3), 67.8 (C-6′), 69.8 (C-5′′), 71.5 (C-4′), 72.1 (C-2′′), 72.4 (C-3′′), 74.1 (C-4′′), 74.9 (C-2′), 76.9 (C-5′), 77.9 (C-3′), 102.1 (C-1′′), 102.5 (C-1′), 100.9 (C-2), 111.6 (C-3), 117.1 (C-4), 139.0 (C-5), 157.1 (C-6), 158.0 (C-1).
References
J. Zhou, R. An, and X. Huang, J. Ethnopharmacol., 270, 113852 (2021).
L. M. Luo, L. Qin, G. Pei, S. G. Huang, X. J. Zhou, and N. H. Chen, Zhongguo Zhong Yao Za Zhi, 43, 1416 (2018).
A. S. Gur′eva, P. K. Kintya, and N. E. Mashchenko, Chem. Nat. Compd., 32, 384 (1996).
J. Yan, W. Guo, L. Zhou, D. Guo, J. Pei, Y. Deng, H. Zheng, D. Liu, X. Xie, and C. Peng, Chem. Biodiv., 18, e2100403 (2021).
S. Hiroko, S. Yutaka, and M. Yoshihiro, Jap. Soc. Pharmacogn., 43, 64 (1989).
I. A. Kim, B. G. Kim, M. Kim, and J. H. Ahn, Phytochemistry, 76, 25 (2012).
X. H. Zhao, D. H. Chen, J. Y. Si, R. L. Pan, and L. G. Shen, Acta Pharm. Sin., 37, 535 (2002).
R. H. Delaporte, K. P. Guzen, A. Laverde, A. R. d. Santos, and M. H. Sarragiotto, Biochem. Syst. Ecol., 34, 599 (2006).
Z.-L. Zhou, S.-Q. Lin, H.-Y. Yang, H.-L. Zhang, and J.-M. Xia, Asian J. Chem., 26, 7616 (2014).
S. Hiroko, S. Yutaka, and M Yoshihiro, Phytochemistry, 27, 451 (1988).
S. J. In, K. H. Seo, N. Y. Song, D. S. Lee, Y. C. Kim, and N. I. Baek, Arch. Pharm. Res., 38, 26 (2015).
T. Tanaka, T. Nakashima, T. Ueda, K. Tomii, and I. Kouno, Chem. Pharm. Bull. (Tokyo), 55, 899 (2007).
Acknowledgment
This research was funded by the 2022 Provincial Science and Technology Research and Development Plan United Fund (222301420075) and the Standardized Preparation and Key Technology Research Projects of Jingfang–Research on Key Techniques for Standardized Preparation of Zhongjing Fang’s “Baihe Dihuang Tang” (GZY-KJS-2022-048-3).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2024, pp. 213–214.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, S., Li, Y., Zhang, M. et al. A New Chlorophenyl Glycoside from the Bulbs of Lilium brownii var. viridulum. Chem Nat Compd 60, 241–243 (2024). https://doi.org/10.1007/s10600-024-04296-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-024-04296-5