
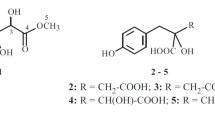
A new coumarin derivative, 7-O-methylparamicoumarin A (1), has been isolated from the roots of Chionanthus retusus, together with seven known compounds. The structure of new compound 1 was determined through spectroscopic and MS analyses. Among the isolates, luteolin (3), quercetin (4), and apigenin (5) exhibited cytotoxicities with IC50 values of 13.69 ± 1.10, 10.41 ± 0.79, and 12.25 ± 0.91 μM, respectively, against DLD-1 cell line. Luteolin (3), quercetin (4), and apigenin (5) also exhibited cytotoxicities with IC50 values of 15.35 ± 1.24, 13.52 ± 0.87, and 19.48 ± 1.26 μM, respectively, against CCRF-CEM cell line. In addition, kaempferol (2), apigenin (5), 3,3′,5,5′,7-pentahydroxyflavanone (6), and oleuropein (8) showed potent inhibition with IC50 values of 24.84 ± 1.95, 27.18 ± 1.82, 25.18 ± 2.07, and 28.14 ± 1.66 μM, respectively, against LPS-induced NO generation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Chionanthus retusus Lindl. & Paxton (Oleaceae) is a deciduous shrub, distributed in China, Korea, Japan, and Taiwan [1]. Previous chemical studies of plants of the genus Chionanthus have reported the isolation of several components including secoiridoids [2], flavonoids [3, 4], lignanoids [3], coumarins [3], and their derivatives. These plants also exhibit diverse biological activities, including antioxidant [2], cytotoxic [3], and neuroprotective [4] activities. The current phytochemical investigation of the roots of C. retusus has led to the isolation of a new coumarin derivative, 7-O-methylparamicoumarin A (1), along with seven known compounds. The structural elucidation of 1 and cytotoxic and anti-inflammatory properties of 1–8 are described herein.
Extensive fractionation of the EtOAc soluble portion of a MeOH extract of roots of Chionanthus retusus using silica gel column chromatography (CC) and preparative TLC afforded compounds 1–8.

7-O-Methylparamicoumarin A (1) was isolated as colorless gum. The HR-ESI-MS gave an [M + Na]+ ion at m/z 351.1575 (calcd for C20H24O4Na, 351.1572), consistent with a molecular formula of C20H24O4. IR absorptions for carbonyl (1710 cm–1) and hydroxyl (3385 cm–1) functions were observed. Comparison of the 1H and 13C NMR data of 1 with those of paramicoumarin A [5] suggested that their structures were closely related, except that the 7-methoxy group [δH 3.99 (3H, s, MeO-7), δC 60.4 (MeO-7)] of 1 replaced the 7-hydroxy group of paramicoumarin A [5]. This was supported by the HMBC correlations observed between MeO-7 (δH 3.99) and C-7 (δ 152.1). The full assignment of 1H and 13C NMR resonances was supported by 1H–1H COSY, DEPT, HSQC, NOESY, and HMBC (Table 1) spectral analyses. On the basis of the above data, the structure of 1 was elucidated as (E)-6-(3,7-dimethylocta-2,6-dien-1-yl)-8-hydroxy-7-methoxy-2H-chromen-2-one and named 7-O-methylparamicoumarin A.
The known isolates were readily identified by a comparison of physical and spectroscopic data (UV, IR, 1H NMR, and MS) with corresponding authentic samples or literature values, and they included kaempferol (2) [3], luteolin (3) [6], quercetin (4) [6], apigenin (5) [3], 3,3′,5,5′,7-penta-hydroxyflavanone (6) [3], phillygenin (7) [3], and oleuropein (8) [3].
Nitric oxide (NO) is a mediator in the inflammatory response involved in host defense. The anti-inflammatory effects of the compounds isolated from the roots of Chionanthus retusus were evaluated by suppressing lipopolysaccharide (LPS)-induced NO generation in murine macrophage cell line RAW264.7. The inhibitory activity data of the isolates 1–8 on NO generation by macrophages are shown in Table 2. Indomethacin was used as a positive control. Among the isolated compounds, kaempferol (2), apigenin (5), 3,3′,5,5′,7-pentahydroxyflavanone (6), and oleuropein (8) showed potent inhibition with IC50 values of 24.84 ± 1.95, 27.18 ± 1.82, 25.18 ± 2.07, and 28.14 ± 1.66 μM, respectively, against LPS-induced NO generation, and were about 5–6 times more effective than indomethacin. Our research indicates that C. retusus and its constituents (especially 2, 5, 6, and 8) deserve further investigation as potential candidates for the treatment or prevention of various inflammatory diseases.
Isolated compounds from the roots of Chionanthus retusus were tested in vitro against DLD-1, CCRF-CEM, HL-60, and IMR-32 cell lines, with cytotoxicity data shown in Table 3. The anticancer agent doxorubicin was used as a positive control. Among the isolates, luteolin (3), quercetin (4), and apigenin (5) exhibited cytotoxicities with IC50 values of 13.69 ± 1.10, 10.41 ± 0.79, and 12.25 ± 0.91 μM, respectively, against DLD-1 cell line. Luteolin (3), quercetin (4), and apigenin (5) also exhibited cytotoxicities with IC50 values of 15.35 ± 1.24, 13.52 ± 0.87, and 19.48 ± 1.26 μM, respectively, against CCRF-CEM cell line. In addition, kaempferol (2) and apigenin (5) showed cytotoxic effects with IC50 values of 19.01 ± 1.23 and 15.66 ± 1.03 μM, respectively, against HL-60 cell line. Previous studies had also shown that these flavonoids (2–5) have anticancer activities against human tumor cell lines (A549, SK-OV-3, SK-MEL-2, and HCT15) [3]. Thus, this suggests that C. retusus and its flavonoid derivatives may be potential candidates for further investigation of various cancer diseases.
Experimental
General Experimental Procedures. UV spectra were obtained on a Jasco UV-240 spectrophotometer. IR spectra (neat or KBr) were recorded on a Perkin Elmer 2000 FT-IR spectrometer. NMR spectra, including COSY, NOESY, HMBC, and HSQC experiments, were recorded on a Varian Inova 500 spectrometer operating at 500 MHz (1H) and 125 MHz (13C), with chemical shifts given in ppm (δ), using tetramethylsilane (TMS) as internal standard. ESI and HR-ESI mass spectra were recorded on a Bruker APEX II mass spectrometer. Silica gel (70–230, 230–400 mesh) (Merck) was used for column chromatography (CC). Silica gel 60 F-254 (Merck) was used for thin-layer chromatography (TLC) and preparative thin-layer chromatography (PTLC). High-performance liquid chromatography (HPLC) was performed using an ODS column (Biosil PRO-ODS-U, 10 mm i.d. × 250 mm; detector, RI).
Plant Material. The roots of Chionanthus retusus were collected from Taoyuan City, Taiwan, in August 2018, and a voucher specimen (CR 201808) was deposited in the Department of Pharmacy, National Yang Ming Chiao Tung University, Taipei, Taiwan.
Extraction and Separation of Compounds. The dried roots (2.1 kg) of C. retusus were extracted three times with MeOH (10 L each) for 3 days. The methanol extract (205 g) was partitioned between EtOAc–H2O (1:1) to afford EtOAc-soluble (Fr. A, 68 g) and H2O-soluble (Fr. B, 136 g) fractions. The EtOAc-soluble fraction (68 g) was chromatographed on silica gel (70–230 mesh, 3.0 kg), eluting with CH2Cl2 and gradually increasing the polarity with MeOH to give 13 subfractions (A1–A13). Subfraction A4 (4.5 g) was separated by column chromatography on silica gel (230–400 mesh, 205 g), eluting with n-hexane–EtOAc (8:1–0:1), to yield 12 subfractions (A4-1–A4-12). Part (140 mg) of Subfr. A4-4 was purified by preparative TLC (silica gel, n-hexane–EtOAc, 6:1) to give 7-O-methylparamicoumarin A (1) (4.1 mg). Part (105 mg) of Subfr. A4-5 was purified by preparative TLC (silica gel, n-hexane–EtOAc, 4:1) to afford phillygenin (7) (6.0 mg). Subfraction A7 (4.8 g) was separated by column chromatography on silica gel (230–400 mesh, 220 g), eluting with CH2Cl2–MeOH (5:1–0:1), to yield 15 subfractions (A7-1–A7-15). Part (130 mg) of Subfr. A7-9 was purified by preparative TLC (silica gel, CH2Cl2–MeOH, 3:1) to afford apigenin (5) (6.5 mg). Subfraction A10 (4.0 g) was separated by column chromatography on silica gel (230–400 mesh, 180 g), eluting with CH2Cl2–MeOH (3:1–0:1), to yield 10 subfractions (A10-1–A10-10). Subfraction A10-5 (350 mg) was purified by MPLC (16 g of SiO2, 230–400 mesh; CH2Cl2–MeOH, 3:1–0:1, 140-mL fractions) to give nine subfractions (A10-5-1–A10-5-9). Subfraction A10-5-5 (40 mg) was purified by HPLC (ODS column, MeOH–H2O, 2:1, 2.0 mL/min) to obtain kaempferol (2) (4.6 mg) and luteolin (3) (5.1 mg). Subfraction A10-5-7 (38 mg) was purified by HPLC (ODS column, MeOH–H2O, 2:1, 2.0 mL/min) to obtain oleuropein (8) (4.5 mg). Subfraction A10-5-8 (42 mg) was purified by HPLC (ODS column, MeOH–H2O, 3:1, 2.0 mL/min) to obtain 3,3′,5,5′,7-pentahydroxyflavanone (6) (4.0 mg). Subfraction A10-7 (320 mg) was purified by MPLC (15 g of SiO2, 230–400 mesh; CH2Cl2–MeOH, 2:1–0:1, 130-mL fractions) to give eight subfractions (A10-7-1–A10-7-8). Subfraction A10-7-6 (37 mg) was purified by HPLC (ODS column, MeOH–H2O, 4:1, 2.0 mL/min) to obtain quercetin (4) (6.5 mg).
7- O -Methylparamicoumarin A (1), colorless gum. UV (MeOH, λmax, nm): 267, 341. IR (neat, νmax, cm–1): 3385 (OH), 1710 (C=O). 1H NMR (500 MHz) and 13C NMR (125 MHz), see Table 1. ESI-MS m/z 351 [M + Na]+. HR-ESI-MS m/z 351.1575 [M + Na]+ (calcd for C20H24O4Na, 351.1572).
Cytotoxic Assay. Cell lines used in this study were DLD-1 cells (human colorectal carcinoma), CCRF-CEM cells (human lymphoblastic leukemia), HL-60 cells (human myeloid leukemia), and IMR-32 cells (human neuroblastoma). The above cell lines were purchased from the Bioresource Collection and Research Center (BCRC), Food Industry Research and Development Institute (FIRDI), Hsinchu 300, Taiwan.
Cytotoxic activities of compounds 1–8 against DLD-1, CCRF-CEM, HL-60, and IMR-32 were assayed by a modification of the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric method [7]. To measure cytotoxic activities of purified compounds against above tumor cells, each cell line was initiated at 1 × 105 cells/well in 96-well microtiter plates (Falcon). Eight concentrations (0.625, 1.25, 2.5, 5, 10, 20, 40, and 80 μM) (triplicate) of test compounds (dissolved in 0.5% DMSO) encompassing a 128-fold range were added to each cell line. Each tumor cell was enumerated using MTT (Sigma) after exposure to test compounds for 3 days; 15 μL of 1 mg/mL MTT was added to each well, and plates were incubated at 37°C for a further 4 h. Formazan crystals were redissolved in DMSO (Merck) for 10 min with shaking, and the plate was read immediately on a microtiter plate reader (Dynatech) at a wavelength of 570 nm. The IC50 value was defined as the concentration of the test compound necessary to inhibit the growth to 50% of the control in the MTT assay. The anticancer agent doxorubicin and 0.5% DMSO were used as positive control and solvent control, respectively. The assays were repeated three times.
Measurement of Nitric Oxide/Nitrite. The murine macrophage cell line RAW264.7 (BCRC No. 60001) was purchased from the Bioresources Collection and Research Center (BCRC, Hsinchu, Taiwan) of the Food Industry Research and Development Institute (Hsinchu, Taiwan). The murine macrophage cell line RAW264.7 was cultured in Dulbecco′s modified Eagle′s medium (DMEM, Gibco BRL Life Technologies, Inc.) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and incubated at 37°C in a humidified 5% CO2 atmosphere with a 96-well flat-bottomed culture plate. After 24 h, the condition medium was replaced with fresh DMEM and FBS. Then compounds 1–8 (2.5, 5, 10, 20, and 40 μM) were added in the presence of lipopolysaccharide (LPS; 1 μg/mL; Sigma, Cat. No. L-2654) and the whole incubated under the same condition for 24 h. The cultured cells were then centrifuged, and the supernatants were used for NO-production measurement. The supernatant was mixed with an equal volume of Griess reagent (1% sulfanilamide, 0.1% N-(naphthalen-1-yl)ethylenediamine dihydrochloride in 2.5% H2PO4 soln.) and incubated for 10 min at room temperature. Nitrite concentration was determined by measuring the absorbance at 540 nm using an ELISA plate reader (μ Quant) [8]. The percentage of NO inhibition of the test compound was calculated as follows:
The data were expressed as a mean of four experiments. The software SigmaPlot was used for determining the IC50 values.
Statistical Analysis. Results are expressed as the mean ± SEM, and comparisons were made using Tukey′s HSD test. A probability of 0.05 or less was considered significant. The software SigmaPlot was used for the statistical analysis.
References
Y. P. Yang and S. Y. Lu, Oleaceae in Flora of Taiwan, Vol. 4, 2nd Ed., Editorial Committee of the Flora of Taiwan, Taipei, Taiwan, 1998, pp. 128–143.
I. Gulcin, R. Elias, A. Gepdiremen, K. Taoubi, and E. Koksal, Wood Sci. Technol., 43, 195 (2009).
J. H. Kwak, M. W. Kang, J. H. Roh, S. U. Choi, and O. P. Zee, Arch. Pharm. Res., 32, 1681 (2009).
Y. G. Lee, H. Lee, J. W. Jung, K. H. Seo, D. Y. Lee, H. G. Kim, J. H. Ko, D. S. Lee, and N. I. Baek, Int. J. Mol. Sci., 20, 3517 (2019).
D. H. Trinh, P. T. Tran, B. T. D. Trinh, H. T. Nguyen, H. D. Nguyen, L. D. Ha, and L. H. D. Nguyen, Phytochem. Lett., 35, 94 (2020).
C. W. Fang, L. C. Chen, H. C. Huang, T. H. Chang, C. L. Chen, M. J. Cheng, and J. J. Chen, Chem. Nat. Compd., 55, 821 (2019).
T. Mosmann, J. Immunol. Methods, 65, 55 (1983).
M. Johansson, B. Kopcke, H. Anke, and O. Sterner, J. Antibiot., 55, 104 (2002).
Acknowledgment
This research was supported by a grant from Ministry of Science and Technology, Taiwan (MOST 109-2320-B-010-029-MY3), awarded to Prof. J.-J. Chen. This work was also supported by the grants from Kaohsiung Veterans General Hospital Tainan Branch (VHYK109-01) and Pingtung Christian Hospital (PS109030). We gratefully thank Ms Shou-Ling Huang and Dr. Iren Wang for assistance in the NMR experiments of the Instrumentation Center at NTU, which is supported by the Ministry of Science and Technology, Taiwan. I-Chou Wang and Li-Chai Chen have contributed equally to this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2021, pp. 716–719.
Rights and permissions
About this article

Cite this article
Wang, IC., Chen, LC., Chang, TH. et al. New Coumarin and Bioactive Constituents of Chionanthus retusus. Chem Nat Compd 57, 835–839 (2021). https://doi.org/10.1007/s10600-021-03492-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-021-03492-x