
Two new oleanane triterpenoids, sysamarins F (1) and G (2), along with one known analogue (3), were isolated from the leaves of Syzygium samarangense. Their structures were elucidated by means of extensive spectroscopic techniques, including interpretation of 1D and 2D NMR spectra and comparison with the values reported in the literature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Syzygium samarangense (Blume) Merr. & L.M. Perry, an evergreen plant, is mainly cultivated in Malaysia, Thailand, Indonesia, and Taiwan [1]. It has been used for the treatment of various diseases such as diabetes, cough, dysentery, inflammation, ringworm, and fever [2,3,4]. Previous phytochemical investigations demonstrated that diversified triterpenoids were one of its main chemical constituents [1, 3,4,5,6]. More recently, we reported on the five new triterpenoids isolated from this plant [7]. In our ongoing search for potential bioactive triterpenoids from S. samarangense, two new oleanane triterpenoids, sysamarins F (1) and G (2), together with one known analogue methyl 2α,3β,23-trihydroxyolean-12-en-28-formate (3), were obtained. We describe herein the isolation and structure elucidation of the new compounds.
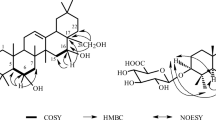
Compound 1 was obtained as a colorless amorphous powder, and its molecular formula was determined as C47H80O6 by the HR-ESI-MS at m/z 763.5824 [M + Na]+ (calcd 763.5847), requiring eight degrees of unsaturation. The 1H NMR spectrum of 1 (Table 1) exhibited eight methyl signals attributable to six tertiary methyl signals (δ 1.18, 1.04, 0.93, 0.91, 0.78, and 0.76), an oxymethyl signal (δ 3.61), and one terminal methyl signal (δ 0.88, t, J = 7.2 Hz), together with an olefinic methine signal (δ 5.27, t, J = 3.6 Hz), an oxymethylene signal (δ 3.96, 3.94, d, J = 10.8 Hz), and two oxymethine signals (δ 3.68, overlapped; 3.28, dd, J = 9.6, 3.6 Hz). Additionally, two ester carbonyl signals (δ 178.1, 173.5) and two olefinic carbons (δ 144.9, 123.1) were observed in the 13C NMR spectrum (Table 1). Further analysis of the NMR data of compound 1 showed high similarities to 3, with the only difference being that a hydroxyl at C-23 in 3 was replaced by a long-chain aliphatic ester moiety (δH 2.32, 1.62, 1.28–1.33 × 10, 1.29, 1.29, 0.88; δC 173.5, 35.1, 31.4, 31.4, 30.5, 30.5, 30.4, 30.4, 30.3, 30.3, 30.2, 30.1, 30.1, 26.1, 23.1, 14.5), as verified by the HMBC correlations from H2-23 (δ 3.96 and 3.94) to the carbonyl (δ 173.5) embedding in the aliphatic chain (Fig. 1).

Key 2D NMR correlations of 1.

Furthermore, two hydroxyls were located at C-2 and C-3 based on the 1H–1H COSY correlations of H-1/H-2/H-3, as well as the long-range HMBC couplings of H-2/C-1 (δ 47.5) and C-4 (δ 42.9) and H-3/C-5 (δ 48.4) and C-24 (δ 13.8), respectively. The HMBC correlations from OMe (δH 3.61) to C-28 (δ 178.1) disclosed that the C-28 carboxyl group was methyl-esterified (Fig. 1). The relative configuration of 1 was determined by means of NOESY experiments, in which cross peaks of H-2/CH3-25, of H-3/H-5 and H2-23, and of H-5/H2-23 indicated that 2-OH, 3-OH, and CH2-23 were assigned α, β, and α, respectively (Fig. 1). Hence, compound 1 was elucidated as methyl 2α,3β-dihydroxy-23α-palmitoyloxyolean-12-en-28-formate and named sysamarin F.
Compound 2 is a colorless amorphous powder; its HR-ESI-MS data at m/z 827.6409 [M + COOH]– (calcd 827.6406) afforded its molecular formula of C50H86O6. Detailed comparison of NMR data of 2 with those of compound 1 disclosed that 2 was also an oleanane triterpenoid, the only difference being the length of the aliphatic ester chain at C-23 between compounds 2 and 1.
The aliphatic ester chain of 2 was deduced to be C19H37O2 fixing at C-23, as evidenced by the 13C NMR (Table 2), HR-ESI-MS, and 2D NMR spectra. Therefore, compound 2, named sysamarin G, was assigned as methyl 2α,3β-dihydroxy-23α-nonadecanoyloxyolean-12-en-28-formate.
In summary, two new oleanane triterpenoids and one known analogue were isolated from the leaves of S. samarangense (Blume) Merr. & L.M. Perry collected from Xishuang Banna Prefecture, Yunnan Province, China. To the best of our knowledge, the oleanane triterpenoids with a long aliphatic chain at C-23 were firstly obtained by us from the genus Syzygium, which not only provided new evidences for the chemical diversity of Syzygium plants but may also be potential chemotaxonomic markers for this species.
EXPERIMENTAL
General. Optical rotations were measured on a JASCO DIP-360 digital polarimeter using 10-cm cell tube. UV spectra were acquired in MeOH with a Shimadzu UV-2401PC UV-vis spectrophotometer. IR spectra were measured on a Bruker Tensor 27 FTIR spectrometer with KBr disks. NMR spectra were recorded in acetone-d6 using a Bruker Avance III-600 spectrometer, and TMS was used as internal standard. HR-ESI-MS data were obtained using an Agilent G6230 Q-TOF mass instrument. Column chromatography (CC) was performed using silica gel (100 × 200 mesh and 200 × 300 mesh, Qingdao Marine Chemical Inc., China) and Sephadex LH-20 (25 × 100 μm, Pharmacia Biotech Ltd., Sweden). Thin-layer chromatography (TLC) was performed using precoated silica gel GF254 plates (Qingdao Marine Chemical Inc., China) with various solvent systems. Semipreparative HPLC was performed on a Hitachi Chromaster system (Hitachi, Ltd., Japan) equipped with a YMC-Triart C18 column (250 mm × 10 mm i.d., 5 μm, YMC Corporation, Japan) using a flow rate of 3.0 mL/min at a column temperature of 25°C, and detection was performed with a DAD detector.
Plant Material. The leaves of Syzygium samarangense (Blume) Merr. & L.M. Perry were collected in September 2012 from Xishuang Banna Tropical Botanical Garden, Yunnan Province, People′s Republic of China, and authenticated by Yu Chen, Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (No. Chen 20120927) was deposited at Kunming Institute of Botany, Chinese Academy of Sciences.
Extraction and Isolation. The air-dried and powdered leaves of S. samarangense (5 kg) were extracted with MeOH (3 × 15 L) at room temperature. The solvent was concentrated under reduced pressure to give a crude extract (0.95 kg). The MeOH extract was then suspended in H2O (1 L) and successively partitioned with petroleum ether (4 × 2 L), EtOAc (4 × 2 L) and n-BuOH (4 × 2L) to yield three parts. The EtOAc extract (112 g) was chromatographed on a silica gel column eluting with a step gradient of petroleum ether–acetone (100:1 to 0:1) to afford 14 fractions (Frs. A–N) based on TLC analysis.
Fraction I (8.1 g) was subjected to CC over silica gel eluting with petroleum ether–EtOAc (7:1 to 1:1) to yield three subfractions (Subfrs. I-1–3). Subfraction I-3 (0.4 g) was separated on a Sephadex LH-20 column (MeOH–CHCl3, 1:1) to give three subfractions (Subfrs. I-3-1–3). Subfraction I-3-2 (171 mg) was chromatographed on a silica gel column using CHCl3–acetone (7:1 to 1:0), then separated over a Sephadex LH-20 column eluting with (MeOH–CHCl3, 1:1), followed by semipreparative HPLC eluting with MeOH–H2O (99:1) to obtain compounds 1 (4.5 mg, tR = 28.3 min) and 2 (1.0 mg, tR = 29.2 min). Fraction J (7.6 g) was chromatographed on a Sephadex LH-20 column eluting with MeOH to get two subfractions (Subfrs. J-1–2) based on TLC analysis. Subfraction J-1 (1.2 g) was subjected to column chromatography (CC) on silica gel (200–300 mesh) eluting with CHCl3–acetone (40:1 to 10:1) to afford two subfractions (Subfrs. J-1-1–2) based on TLC analysis. Subfraction J-1-1 (184 mg) was purified on a Sephadex LH-20 column (MeOH–CHCl3, 1:1) followed by semipreparative HPLC (MeOH–H2O, 39:11) to yield compound 3 (25.0 mg, tR = 9.6 min).
Sysamarin F (1), \( {\left[\upalpha \right]}_{\mathrm{D}}^{21.8} \) +42.7° (c 0.2, MeOH). UV (MeOH, λmax, nm) (log ε): 205 (3.07). IR (KBr, ν, cm–1): 3428, 2925, 2854, 1734, 1632, 1465, 1385, 1260, 1210, 1164, 1124, 1093, 1034, 967, 873, 805, 721. HR-ESI-MS m/z 763.5824 [M + Na]+ (calcd for C47H80O6Na, 763.5847). For 1H and 13C NMR data, see Table 1.
Sysamarin G (2), \( {\left[\upalpha \right]}_{\mathrm{D}}^{21.4} \) +28.4° (c 0.1, MeOH). UV (MeOH, λmax, nm) (log ε): 204 (3.14). IR (KBr, ν, cm–1): 3426, 2925, 2854, 1734, 1631, 1465, 1385, 1365, 1260, 1210, 1164, 1124, 1095, 1035, 967, 873, 805, 721. HR-ESI-MS (negative mode) m/z 827.6409 [M + COOH]– (calcd for C50H86O6COO–, 827.6406). For 1H and 13C NMR data, see Table 2. The structure of 3 was identified as methyl 2α,3β,23-trihydroxyolean-12-en-28-formate by comparing their spectroscopic data with the literature values [8].
References
M. N. Samy, S. Sugimoto, K. Matsunami, H. Otsuka, and M. S Kamel, Chem. Pharm. Bull., 62, 1013 (2014).
Y. Kuo, L. Yang, and L. Lin, Planta Med., 70, 1237 (2004).
C. Ragasa, F. F. JR, D. Raga, and C. C. Shen, Pharm. Chem., 6, 256 (2014).
Y. Z. Wu, Y. B. Zhang, N. H. Chen, G. C. Wang, and Y. L. Li, Zhongyaocai, 38, 754 (2015).
R. Srivastava, A. K. Shaw, and D. K. K. Kulshreshtha, Phytochemistry, 38, 687 (1995).
D. D. Raga, C. L. Cheng, K. C. Lee, W. Z. Olaziman, V. J. De Guzman, C. C. Shen, F. C. Franco, and C. Y. Jr Ragasa, Z. Naturforsch C, 66, 235 (2011).
Y. K. Hu, L. Wang, Y. Y. Li, M. J. Li, W. Xu, Y. Zhao, F. Li, and Y. Zhao, Phytochem. Lett., 25, 147 (2018).
M. W. Xia, J. J. Tan, L. Yang, Z. P. Shang, Q. C. Zhao, and G. B. Shi, Chin. Trad. Herb. Drug, 41, 1612 (2010).
ACKNOWLEDGMENT
This work was financially supported by the National Natural Science Foundation of China (No. 31560098).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 4, July–August, 2020, pp. 598–600.
Rights and permissions
About this article

Cite this article
Hu, YK., Wang, L., Zhao, Y. et al. Two New Oleanane Triterpenoids from Syzygium samarangense. Chem Nat Compd 56, 692–695 (2020). https://doi.org/10.1007/s10600-020-03121-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-020-03121-z