
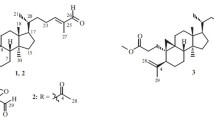
A new cycloartane-type triterpenoid, 24,31-epoxy-24-ethylcycloartan-3α-ol (1), was isolated from the rhizomes of Polygonum bistorta. The structure of 1 was elucidated using a combination of 1D and 2D NMR spectroscopic techniques and HR-EI-MS analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Polygonum bistorta belongs to the Polygonaceae family [1,2,3]. P. bistorta has been used in traditional Chinese, Japanese, and Indian medicines as a remedy for pimples, jaundice, smallpox, measles, insect stings, snake bites, and expelling worms [3]. P. bistorta also finds its application in the treatment of a wide range of complaints including cystitis, irritable bowel syndrome, ulcerative colitis, peptic ulcers, dysentery, diarrhea, cholera, etc. [3]. P. bistorta is one of the strongest herbal astringents [1, 2]. The roots and leaves are either cooked or eaten as raw food in America and Europe [4, 5]. The antibacterial [6, 7], antifungal [8], antioxidant [8, 9], anti-mutagenicity [10], anti-inflammatory [11, 12], and cytotoxic [2, 13] activities of P. bistorta have been reported previously. Triterpenoids [1, 2, 14], cycloartane type triterpenoids [1, 2], steroids [1, 2, 14], tannins [7], flavonoids [1, 2, 15], and phenolics [16, 17] have been isolated and identified as active ingredients from P. bistorta. In this paper, we report a new cycloartane-type triterpenoid, viz. 24,31-epoxy-24-ethylcycloartan-3α-ol (1), from the rhizomes of P. bistorta.

24,31-Epoxy-24-ethylcycloartan-3α-ol (1) was obtained as a colorless powder. It gave a molecular ion peak at m/z 470.4108 in the HR-EI-MS, and therefore its molecular formula has been deduced as C32H54O2. Inspection of its 13C NMR and HSQC-DEPT spectra revealed the presence of 32 signals: eight methyl, eleven methylene, seven methine, and six quaternary carbons.
The presence of a cycloartane skeleton in compound 1 was identified through its 1H NMR spectrum with typical high-field AB doublets due to the presence of non-equivalent methylene protons at C-19 with chemical shift values of δ 0.51 (1H, d, J = 4.2 Hz, H-19α) and δ 0.36 (1H, d, J = 4.2 Hz, H-19β) [1, 18, 19] (Table 1). Additionally, a fragment ion at m/z 315 [M – C10H19O]+ was observed in the EI-MS spectrum, which was also evidence of the presence of a cycloartane skeleton having a hydroxyl group in the nucleus and a C10 side chain [1, 19].
The 1H–1H COSY spectrum together with HMBC data revealed that compound 1 has seven distinct 1H–1H spin systems, viz. (a) [-CH2-CH2-CH-(O)-], (b) [>CH-CH2-CH2-CH<], (c) [>CH2-CH2-], (d) [-CH2-CH2-CH<], (e) [-CH-(CH3)- CH2-CH2-], (f) [-CH-(CH3)2], and (g) [-CH3-CH-(O)-]. The spin system (a) [-CH2-CH2-CH-(O)-] was assigned for C-1, C-2, and C-3. In the HMBC spectrum, C-1 (δ 27.3) showed correlation with H-2, C-2 (δ 28.4) showed correlation with H-1, and C-3 (δ 77.2) showed correlations with H-2, H-28, and H-29. These correlations together with the 13C NMR chemical shift value at C-3 (δ 77.2) and the 1H NMR chemical shift value at H-3 (δ 3.50, 1H, br.s) allowed us to place one of the oxymethine protons at C-3. In other words, the position of the hydroxyl group was fixed at C-3. C-4 (δ 39.7) is a quaternary carbon that showed correlations with methyl protons at H-28 (δ 0.90, 3H, s) and H-29 (δ 0.92, 3H, s), and these correlations allowed us to place the two methyl groups (belonging to C-28, δ 22.4 and C-29, δ 20.8) at the same carbon C-4. The spin system (b) [>CH-CH2-CH2-CH<] was assigned for the positions C-5 to C-8. C-5 (δ 41.0) showed correlation with H-19, and C-6 (δ 21.2) showed correlation with H-5. H-6 protons resonated at δ 1.60 (1H, m, H-6α) and δ 0.83 (1H, m, H-6β), and this multiplicity indicated that each proton showed vicinal couplings with H-7 and H-5 and geminal coupling with H-6.
Similarly, C-7 (δ 28.2) showed correlation with H-6, and C-8 (δ 48.0) showed correlations with H-19 and H-30. H-8 resonated at δ 1.47 (1H, dd, J = 12.5, 4.9 Hz), and this multiplicity was due to the vicinal coupling with H-7. All these correlations are in good agreement with the above spin system and allowed us to form a skeleton of C-5 to C-8. C-9 (δ 19.8) and C-10 (δ 26.4) are quaternary carbons that showed correlation with H-1 and H-6, respectively. The spin system (c) [>CH2-CH2-] was assigned for the positions C-11 and C-12 since C-11 (δ 26.2) showed correlations with H-8 and H-19, and C-12 (δ 32.8) showed correlation with H-18. C-13 (δ 45.5) is a quaternary carbon that showed correlations with H-8 and H-18. Additionally, these correlations allowed us to place the methyl group belonging to C-18 (δ 19.5) at C-13. C-14 (δ 49.2) is also a quaternary carbon that showed correlation with H-18. C-15 (δ 35.6) showed correlations with H-30, and C-8 (δ 48.0) showed correlation with H-30, and therefore the methyl group belonging to C-30 (δ 19.5) was placed at C-14 (δ 49.2). The spin system (d) [-CH2-CH2-CH<] was assigned for positions C-15 to C-17. C-15 (δ 35.6) showed correlations with H-16 and H-30, and C-17 (δ 52.2) showed correlation with H-21.
C-19 (δ 29.7) was identified as a methylene group belonging to the cyclopropane ring. The H-1 (δ 1.57, 1H, m, H-1α and δ 1.25, m, H-1β) and H-11 (δ 1.90, 1H, m, H-11α and δ 1.12, 1H, m, H-11β) protons showed correlations with carbon at C-19 (δ 29.7) and C-5 (δ 41.0), and C-8 (δ 48.0) showed correlation with H-19. These correlations allowed us to place this methylene group between C-9 (δ 19.8) and C-10 (δ 26.4). Further, H-19 protons resonated at high field at δ 0.51 (1H, d, J = 4.2 Hz, H-19α) and δ 0.36 (1H, d, J = 4.2 Hz, H-19β); they are nonequivalent, and each proton splits into a doublet due to geminal coupling with each other. The spin system (e) [-CH-(CH3)-CH2-CH2-] was assigned for the positions C-20 to C-23. C-20 (δ 36.1) showed correlation with methyl protons at H-21; this methyl proton splits into a doublet (3H, δ 0.95, d, J = 5.0 Hz, H-21) due to vicinal coupling with H-20. C-21 (18.3 ppm) showed correlation with H-17, and C-22 (δ 34.8) showed correlation with H-21. C-24 (δ 65.2) is a quaternary carbon. The spin system (f) [-CH-(CH3)2] was assigned for C-25 (δ 29.8), C-26 (δ 21.9), and C-27 (δ 21.9), i.e., for an isopropyl moiety at C-24. In the HMBC spectrum, C-25 (δ 29.8) showed correlations with H-26 and H-27, and C-24 (δ 65.2) showed correlations with H-23, H-25, H-26, and H-27. Each of the methyl protons at H-26 and H-27 splits into a doublet due to vicinal coupling with H-25. All these correlations indicated the presence of an isopropyl moiety at C-24 (δ 65.2). Finally, the spin system (g) [-CH3-CH-] was assigned for C-31 and C-32. C-31 (δ 60.2) showed correlations with H-23, H-25, and H-32. Similarly, C-32 (δ 12.8) showed correlation with H-31. Further, C-24 (δ 65.2) showed correlations with H-31 and H-32. In the 1H NMR spectrum, the protons in the methyl group at C-32 gave a doublet at δ 1.58 (3H, d, J = 6.2 Hz, H-32) due to vicinal coupling with H-31. The single proton on C-31 clearly showed the expected quartet at δ 2.87 (1H, q, J = 6.5 Hz, H-31) due to vicinal coupling with H-32. All these correlations indicated the presence of a [CH3-CH<] moiety at C-24.
The 1H NMR chemical shift value at the C-3 position was observed at δ 3.50 (1H, br.s, H-3), and this value also supported the presence of an oxygenated carbon at C-3. Additionally, these chemical shift values indicated that the orientation of the hydroxyl group at this position was α; this was confirmed by comparing their chemical shift values with closely related compounds such as 24(E)-ethylidenecycloartan-3α-ol etc. [1, 20]. For a β-orientation, these chemical shift values would be around δ 3.28 (m) and δ 78.8 [21, 22]. Two other oxygenated carbons were assigned to C-24 (δ 65.2) and C-31 (δ 60.2), respectively, based on their chemical shift values (Table 1). In addition, the HSQC and DEPT spectrum indicated that C-24 and C-31 were respectively quaternary and methine carbons. Based on these observations, we consider the oxygen atom as an epoxy function between the C-24 and C-31 positions, unambiguously. This fact has further been supported by the presence of a fragment ion in the mass spectrum at m/z 454 [M – O]+. Based on the above interpretation, the structure of the new compound 1 has been elucidated as 24,31-epoxy-24-ethylcycloartan-3α-ol 1. The MS pattern of major fragment ions of 24,31-epoxy-24- ethylcycloartan-3α-ol 1 is given in Fig. 1.

MS pattern of major fragment ions of 24,31-epoxy-24-ethylcycloartan-3α-ol (1).
Experimental
General Experimental Procedures. 1D and 2D NMR spectra were recorded on a Bruker 500 MHz spectrometer, with TMS as a reference standard. LR-EI-MS and HR-EI-MS were measured on Finnigan/MAT MAT 95 XL-T mass spectrometers. Silica gel 60 (Merck, 0.063–0.200 m) was used for column chromatography. Precoated silica gel plates (Merck, Kieselgel 60F 254, 0.25 mm or Baker Si250F, 0.25 mm) were used for preparative TLC and/or analytical TLC. Lichroprep RP-18 (Merck, 40–63 μm) was used for separation and/or purification. Spots were detected using UV light or staining with iodine crystals or by spraying with 50% H2SO4 and heating at 110°C for 5 min.
Plant Material. The plant materials were purchased from a local market, and a voucher specimen (KMano PB 2003) was deposited in the Department of Biological Sciences, National University of Singapore, Republic of Singapore.
Extraction and Isolation. The rhizomes of P. bistorta (12 kg) were ground into powder and then extracted with chloroform at room temperature. The residue was dissolved in a water–methanol mixture (95:5) and then extracted successively with n-hexane and chloroform. The hexane fraction was chromatographed over a silica gel column using hexane and then eluted in a gradient fashion with solvents of increasing polarity. The chloroform fraction was chromatographed over Lichroprep RP-18 and eluted in isocratic fashion with methanol. Several subfractions were obtained from both hexane and chloroform fractions [1, 2]. These subfractions have been evaluated for their cytotoxic activity against cancer cell lines in culture [2], and subsequently several known and new compounds have been reported [1]. Purification of a minor subfraction by preparative TLC using a solvent system (methanol–chloroform, 9:1) afforded the new cycloartane type triterpenoid, viz. 24,31-epoxy-24-ethylcycloartan-3α-ol 1 (ca. 1.0 mg).
24,31-Epoxy-24-ethylcycloartan-3α-ol (1). Colorless amorphous powder. 1H NMR (500 MHz, CDCl3, δ, ppm, J/Hz) and 13C NMR (125 MHz, CDCl3, δ, ppm), see Table 1. MS (EI, 70eV), m/z (Irel., %): 470 (M+, 8), 454 (48), 439 (64), 356 (33), 315 (45), 271 (44), 201 (78), 175 (92), 95 (100), 55 (60). HR-EI-MS m/z 470.4108 (calcd for C32H54O2, 470.4123).
References
M. K. Pillai, T. K. H. Benny, and Y. Daiwen, Phytochemistry, 66 (19), 2304 (2005).
M. K. Pillai, Y. Daiwen, H. Annie, and T. K. H. Benny, Med. Chem., 3 (2), 121 (2007).
A. Intisar, L. Zhang, H. Luo, J. B. Kiazolu, R. Zhang, and W. Zhang, Afr. J. Trad. Complement Altern. Med., 10 (1), 53 (2013).
F. Couplan and J. A. Duke, The Encyclopaedia of Edible Plants of North America, Keats Publishing Inc., New Canaan, Connecticut, 1998, p.128.
D. E. Moerman, Native American Food Plants: an Ethnobotanical Dictionary, Timber Press Inc., Portland, Oregon, 1998, p. 189.
A. Khalid, A. Waseem, M. Saadullah, U-U. Rehman, S. Khiljee, A. Sethi, M. H. H. B. Asad, F. Rasool, M. K. Waqas, and G. Murtaza, Afr. J. Pharm. Pharmacol., 5 (7), 887 (2011).
C. Q. Liu, X. L. Wang, and J. Zeng, J. Gannan Med. Univ., 26 (4), 489 (2006).
M. Neelma, I. Wasqa, A. Imaran, and N. Shagufta, Asian Pac. J. Trop. Biomed., 4 (2), S639 (2014).
X. Chang, Y. X. Liu, and W. Y. Kang, Fine Chem. Interm., 39, 28 (2009).
N. Miki, W. A-Fu, S. Takahiko, N. Hisamitsu, and K. Hideaki, Nad. Med., 49, 329 (1995).
M. Duwiejua, I. L. Zeitin, A. I. Gray, and P. G. Waterman, J. Pharm. Pharmacol., 46, 286 (1994).
M. Duwiejua, I. L. Zeitin, A. I. Gray, and P. G. Waterman, Planta Med., 65, 371 (1999).
Y. H. Liu, Y. P. Weng, H. Y. Lin, S. W. Tang, C. J. Chen, C. J. Liang, C. Y. Ku, and J. Y. Lin, Sci. Rep., 7 (1), 41437 (2017).
X. B. Sun, P. H. Zhao, Y. J. Xu, L. M. Sun, M. A. Cao, and C. S. Yuan, Chem. Nat. Compd., 43, 563 (2007).
H. D. Smolarz, Acta Pol. Pharm. Drug Res., 59 (2), 145 (2002).
X. Q. Liu, F. K. Chen, L. J. Wu, S. T. Wang, and W. W. Li, J. Shenyang Pharm. Univ., 3, 187 (2004).
A. Intisar, J. B. Kiazolu, Y. Wang, L. Zhang, and W. Zhang, J. Liq. Chromatogr. Relat. Technol., 35 (7), 977 (2012).
C. Djerassi and R. McCrindle, J. Chem. Soc., 4034 (1962).
Z. Cantillo-Ciau, W. Brito-Loeza, and L. Quijano, J. Nat. Prod., 64, 953 (2001).
A. H. Januario, M. Fatima Das, G. F. Da Silva, P. C. Vieira, and J. B. Fernandes, Phytochemistry, 31, 1251 (1992).
M. D. Greca, A. Fiorentino, P. Monaco, and L. Previtera, Phytochemistry, 35, 1017 (1994).
D. P. J. Teres, J. G. Urones, I. S. Marcos, P. Basabe, C. M. J. Sexmero, and F. Moro, Phytochemistry, 26, 1767 (1987).
Acknowledgment
This research was supported by a grant from the National University of Singapore. One of the authors, Manoharan Karuppiah Pillai, thanks the National University of Singapore for providing financial assistance in the form of a Ph.D. graduate fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 6, November–December, 2019, pp. 934–937.
Rights and permissions
About this article

Cite this article
Pillai, M.K., Huat, B.T.K. & Yang, D. A New Cycloartane-Type Triterpenoid from Polygonum bistorta. Chem Nat Compd 55, 1085–1089 (2019). https://doi.org/10.1007/s10600-019-02900-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-019-02900-7