
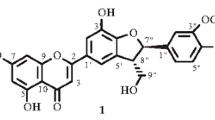
The new flavanonol glycoside (2S,3S)-3,5-dihydroxy-2-(4′-hydroxyphenyl)-8-(3-methylbut-2-en-1-yl)-4-oxo-3,4-dihydro-2H-chromen-7-yl-β-D-glucopyranosyl-(3→1)-xylopyranoside (lavaloside) (1) and the known compound phellamurin (2) were isolated from leaves of Phellodendron lavallei. The structures of the flavanone glycosides were elucidated using chemical transformations and UV, PMR, and 13C NMR spectroscopy combined with HSQC, HMBC, COSY, and DEPT experiments and mass spectroscopy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Phellodendron lavallei Dode (Rutaceae L.) is known to contain various classes of biologically active compounds, e.g., alkaloids, coumarins, and flavonoids. It is used for infectious diseases, pneumonia, tuberculosis, pleurisy, edema, hepatitis, herpes virus, etc. [1,2,3,4].
The chemistry (contents of phenolic compounds) and pharmacology of the plant introduced to the Black Sea shores of Georgia have not been studied. Preliminary chemical analysis of the leaves found significant quantities of alkaloids, phenolic compounds, and polysaccharides [5].
The aqueous extract of air-dried leaves isolated purified total polysaccharides (18% yield); aqueous EtOH extract, total phenolic compounds (5% yield of air-dried raw material). Column chromatography of the latter isolated 1 and 2, the dominant glycosides in it.

Compound 1 was isolated as yellow crystals that gave a positive Bryant cyanidin reaction [6]. The molecular formula C31H38O15 was established using HR-TOF-MS (m/z 651 [M + H]+). UV absorption maxima occurred at 328 and 285 nm. Chemical shifts of H-2 (δ 5.00 ppm, J = 11.5 Hz) and H-3 proton resonances (δ 4.59, J = 11.5 Hz) in the PMR spectrum confirmed that this compound was a flavanonol [7].
Complete assignment of resonances in PMR and 13C NMR spectra (Table 1) and a comparison with 2D NMR experiments showed that 1 contained an isoprenyl chain consisting of five C atoms with chemical shifts δ 17.9, 22.2, 25.6, 123.5, and 131.1. The molecule of 1 also contained two carbohydrates according to resonances at δ 5.05 (d, J = 8.0 Hz) and δ 4.72 (d, J = 7.8 Hz). The chemical shift of β-D-glucose C-3′′′ at δ 87.1 signified that it was substituted.
The spectral data were confirmed by results from chemical conversions. Acid hydrolysis of 1 produced compound 3, D-glucose, and D-xylose. The yield of aglycon (49%) confirmed that the glycoside was a bioside. Stepwise hydrolysis of 1 gave D-xylose and monoglycoside 4, the chromatographic behavior of which was identical to that of 2. Therefore, D-xylose was the terminal sugar whereas D-glucose was bonded directly to the aglycon.
The structure of 1 was elucidated by comparing 2 and the product of stepwise acid hydrolysis of 1 (4). Acid hydrolysis of 4 and 2 produced in both instances a sugar (D-glucose) and genins (3 and 5) that turned out to have identical chemical reactions and PC and TLC mobilities in different solvent systems. However, the hydrolysis product of 4 was yellow (3); of 2, white (5).
Next, aglycons 3 and 5 were oxidized and dealkylated, monitoring their changes in the reaction mixture. Work up of the corresponding oxidized products (6) separated a yellow precipitate 7, mp 275–280°C, C15H10O6. Its UV spectrum was consistent with free hydroxyls on C-3, C-5, C-7, and C-4′, in contrast with scutellarein and luteolin, which have atomic compositions identical to kaempferol. Compound 7 was identified as kaempferol [8].
Products obtained via hydrogenation of 3 and 5 and subsequent alkaline cleavage gave isovaleric acid (8). This was also confirmed by PMR and 13C NMR spectra, which showed an isoprenyl chain in the studied glycosides. A resonance at δ 110.7 in the 13C NMR spectrum indicated that it was located on C-8 in both flavanonols and not on C-6 (chemical shift δ 104 ppm) [3].
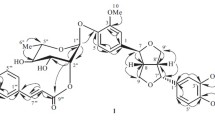
Correlations of atoms in 1 were established using COSY, HSQC, and HMBC spectra (Fig. 1). The HMBC spectrum indicated a correlation between an anomeric proton at δ 5.05 (D-Glc H-1) and C-7 of the aglycon with chemical shift δ 164.4. Therefore, the carbohydrates were probably located on C-7 of genin 3.

HMBC correlations for 1.
A correlation between D-xylose H-1 and D-glucose C-3 provided proof of a D-xylose-(1→3)-D-glucose bond.
According to the literature, flavanonols with 2S,3S-trans-configurations are mainly yellow; with 2R,3R-transconfigurations, white [9, 10]. The choice of the two configurations was made based on the literature.
An analysis of the experimental results and a comparison with the literature characterized 1 as (2S,3S)-3,5-dihydroxy-2-(4′-hydroxyphenyl)-8-(3-methylbut-2-en-1-yl)-4-oxo-3,4-dihydro-2H-chromen-7-yl-β-D-glucopyranosyl-(3→1)-xylopyranoside. A compound with an analogous structure has not been reported. Therefore, it was new and was called lavaloside.
Compound 2 was isolated as white crystals that gave a positive Bryant cyanidin reaction [6]. The mass spectrum with m/z 518 [M + H]+ agreed with the formula C26H29O11. Absorption maxima at 345 and 290 nm in the UV spectrum and chemical shifts for H-2 and H-3 resonances in the PMR spectrum confirmed that it was a flavanonol [7].
Compound 2 was cleaved in acidic solution to form aglycon 5 and D-glucose. Correspondingly, the PMR spectrum had a resonance for one anomeric proton at δ 5.03 (d, J = 8.0 Hz). Resonances for the other D-glucose protons appeared in the range δ 3.42–3.90 (Table 1).
The weak-field part of the spectrum showed resonances (AA′BB′ system) for H-2′, H-6′ and H-3′, H-5′ with
SSCC 8.5 Hz at δ 7.35 and 6.82, respectively, and a 1H resonance for H-6 at δ 6.35. H-2 and H-3 resonated at δ 4.99 and 4.58 as doublets characteristic of an AB system and were confirmed by double resonance with SSCC 11.5 Hz. This was indicative of a 2R,3R-trans-configuration [3].
Compound 2 was identified by the analyses as 2R,3R-3,5-dihydroxy-2-(4′-hydroxyphenyl)-8-(3-methylbut-2-en-1-yl)-4-oxo-3,4-dihydro-2H-chromen-7-yl-β-D-glucopyranoside (phellamurin).
Experimental
IR spectra were recorded on a UR-10 spectrometer; UV spectra, on an SF-16 spectrophotometer. PMR and 13C NMR spectra were recorded in CD3OD on a Bruker Avance DRX 500 FT. 2D NMR spectra used standard Bruker pulse programs. FAB-MS were measured using an E. I. Finnigan Model 4600 Quadrupole System spectrometer. TLC and PC used solvent systems n-BuOH–AcOH–H2O (4:1:2, 1); CHCl3–MeOH–H2O (26:14:3, 2); CHCl3–MeOH (10:1, 3); Py–C6H6–BuOH–H2O (3:1:5:3, 4); and EtOH–H2O–NH4OH (25%) (8:1:1, 5) and detectors MeOH with base (10%, 1); anilinium phthalate (2), and bromothymol blue solution (3). Flavonoids in chromatograms were detected in UV light by reagent 1; sugars, 2; and isovaleric acid, 3. Compounds were separated from the total over a column of silica gel (L 100/160) with elution by CHCl3 and CHCl3–MeOH mixtures with increasing concentration of the latter.
Plant Material. Leaves of P. lavallei were collected in September on the Adjara shores (Georgia) of the Black Sea.
Extraction and Isolation. Air-dried ground leaves (2 kg) were extracted with EtOH (80%, 1:10). The alcohol was evaporated from the extract. The aqueous residue was purified using CHCl3 (3 × 200 mL). The purified aqueous residue was extracted with EtOAc (5 × 300 mL). The EtOAc extract was dried and evaporated to produce a light-yellow amorphous powder (5% yield from air-dried raw material). The total EtOAc extract was separated over a column of silica gel with elution by CHCl3 and CHCl3–MeOH (10:1) with increasing concentration of the latter to isolate 1 and 2.
Lavaloside (1). Yellow crystals, mp 165–167°C (EtOH). IR spectrum (KBr, νmax, cm–1): 3400, 2914, 1640, 1583, 1234. UV spectrum (C2H5OH, λmax, nm): 328, 285, 215 sh. Mass spectrum, m/z: 651 [M + H]+, 519 [M + H – 132]+, 357 [M + H – 132 – 162]+. For 1H and 13C NMR spectral data, see Table 1.
Acid Hydrolysis of 1. Glycoside 1 (0.5 g) was treated with H2SO4 (5 mL, 5%) and heated on a water bath for 1 h. The resulting precipitate was filtered off. The filtrate was neutralized over anion-exchanger AV-17 (OH-form), condensed, and chromatographed on paper using systems 1 and 4. The hydrolysate contained D-glucose and D-xylose. The precipitate on the filter paper was rinsed with H2O, dried in air, and recrystallized to afford 3 (0.23 g), yellow crystals, mp 217–220°C. UV spectrum (EtOH, λmax, nm): 270, 335.
Stepwise Acid Hydrolysis. Lavaloside (0.5 g) was dissolved in EtOH (70 mL, 50%) containing H2SO4 (8%) and hydrolyzed on a water bath. Formation of cleavage products was monitored every 5 min. The amount of intermediate products reached a maximum at 25 min. At this point, the hydrolysis was interrupted. The solution was neutralized with BaCO3 and filtered. The filtrate was evaporated to a small volume. The mixture of flavonoids was extracted with EtOAc. The extract was washed with H2O, dehydrated, and evaporated. The mixture of flavonoids was separated over a column of silica gel with elution by EtOH (45%). The first fractions contained the starting glycoside. An intermediate hydrolysis product crystallized out of subsequent fractions. It was filtered off and dried in air to afford 4 (0.3 g), yellow crystals, mp 220–221°C, that appeared at the level of phellamurin on chromatograms in various solvent systems. UV spectrum (EtOH, λmax, nm): 285, 343. IR spectrum (KBr, νmax, cm–1): 3380 (OH), 2921, 1635 (C=O), 1371, 1074. Table 1 lists the 1H and 13C NMR spectra. PC of the aqueous part of the hydrolysate using system 4 detected D-xylose.
Acid Hydrolysis of 4. Compound 4 (0.05 g) was hydrolyzed analogously to 1 to afford the aglycon 3 and D-glucose.
Oxidation of the Aglycon of 1 by H 2 O 2 [11]. A solution of aglycon 3 or 5 (0.05 g) in MeOH (5 mL) was treated with KOH solution (1 mL, 10%) and H2O2 (0.2 mL) and held at 4°C for 3 d. The course of the reaction was monitored by TLC. The solution was diluted with H2O (up to 20 mL) and neutralized with acid to afford oxidation product 6.
Dealkylation of 6 [11]. Compound 6 (0.04 g) was mixed with HI solution (2 mL, spec. grav. 1.7) and liquid phenol (1.6 mL), heated under gentle reflux for 7 h, cooled when the reaction was finished, and poured into sodium thiosulfate solution (60 mL, 20%). The resulting precipitate was filtered off and dissolved in alcohol. The insoluble part was separated. The diluted aqueous filtrate afforded kaempferol (7), C15H10O6, mp 277–278°C [8].
Hydrogenation of Aglycons 3 and 5. Aglycon (0.2 g) was dissolved in EtOH (5 mL) and treated with Pd/C catalyst. H2 was passed through the reaction mixture. The reduction product was extracted with EtOAc. The extract was evaporated to dryness.
Alkaline Cleavage of the Hydrogenation Product [11]. The product obtained by hydrogenation of aglycon 3 or 5 (0.1 g) was dissolved in dilute KOH solution (0.3 g in 0.1 mL H2O), stored for 5 min, cooled, diluted with H2O (10 mL), and neutralized with HCl solution (10%) to pH 5. The cleavage products distilling with steam contained isovaleric acid (8) according to PC with a standard using systems 1 and 5 (detector reagent 3).
Phellamurin (2). White crystalline powder, mp 151–153°C. UV spectrum (EtOH, λmax, nm): 345, 290. The mass spectrum (m/z 518 [M + H]+) corresponded to the formula C26H29O11. Acid hydrolysis cleaved 2 to form the aglycon 5 and D-glucose [10]. Table 1 lists the PMR and 13C NMR spectra.
References
Plant Resources [in Russian], Vol. 4, Nauka, Leningrad, 1988, 357 pp.
C.-H. Leu, C.-Y. Li, X. Yao, and T.-S. Wu, Chem. Pharm. Bull., 54 (9), 1308 (2006).
C.-Y. Chiu, C.-Y. Li, C.-C. Chiu, M. Niwa, S. Kitanaka, A. G. Damu, E.-J. Lee, and T.-S. Wu, Chem. Pharm. Bull., 53 (9), 1118 (2005).
T.-S. Wu, M.-Y. Hsu, P.-C. Kuo, B. Sreenivasulu, A. G. Damu, C.-R. Su, C.-Y. Li, and H.-C. Chang, J. Nat. Prod., 66, 1207 (2003).
M. D. Alania, K. G. Shalashvili, A. J. Bakuridze, and M. G. Sutiashvili, in: Study of Biologically Active Compounds from Plant and Mineral Raw Materials of Georgia. Collection of Scientific Works [in Georgian], 2 (17), 2010, p. 14.
E. F. Bryant, J. Am. Pharm. Assoc. Sci. Ed., 39, 480 (1950).
T. J. Mabry, K. R. Markham, and M. B. Thomas, The Systematic Identification of Flavonoids, Springer–Verlag, Berlin–Heidelberg–New York, 1970, 354 pp.
M. Alania, Advances in the Chemistry of Secondary Metabolites (Flavonoids and Cycloartanes) of Astragalus from Georgia’s Flora [in Russian], Samshoblo, Tbilisi, 2016, 394 pp.
T. G. Sagareishvili and V. G. Tsitsishvili, Chem. Nat. Compd., 42, 419 (2006).
J. Ma, S. H. Jones, and S. M. Hecht, J. Nat. Prod., 68, 115 (2005).
O. I. Shevchuk, N. P. Maksyutina, and V. I. Litvinenko, Chem. Nat. Compd., 4, 66 (1968).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2018, pp. 219–222.
Rights and permissions
About this article

Cite this article
Shalashvili, K.G., Sutiashvili, M.G., Alaniya, M.D. et al. Flavanonol Glycosides from Leaves of Phellodendron lavallei Introduced in Georgia. Chem Nat Compd 54, 263–266 (2018). https://doi.org/10.1007/s10600-018-2319-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-018-2319-x