
A new hirsutinolide-type sesquiterpene lactone, 8α-(2′Z-tigloyloxy)-1α-methoxyhirsutinolide (1), and a new naturally occurring 8α-(2′-hydroxymethylacryloyloxy)-1α-methoxyhirsutinolide-13-O-acetate (2) were isolated from the CHCl3 extract of the aerial parts of Vernonia cinerea (Asteraceae). The structures of the new compounds were determined by 1D and 2D NMR and MS experiments, as well as by comparison of their data with the published values.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The genus Vernonia comprises about one thousand species. The plant Vernonia cinerea Less. (Asteraceae) is distributed mainly in Africa and Southeast Asia. It is commonly used for treatment of various diseases such as asthma, bronchitis, cough, malaria fever, skin, and liver diseases [1,2,3]. There have been phytochemical reports on the diverse compounds from this species, including sesquiterpene lactones [4, 5], flavonoids [6], and triterpenes [7]. Some of the compounds have been shown to have anticancer [8], antimalarial [5], and antifeedant biological activities [9]. In our previous research, we have reported on the sesquiterpene lactones from the aerial parts of V. cinerea, along with their structure–activity relationships and their anti-inflammation and anticancer properties [10, 11]. In our ongoing chemical and biochemical studies on this plant, a new hirsutinolide-type sesquiterpene lactone (1), together with a new naturally occurring compound (2), has been isolated. This paper reports the isolation and structure elucidation of the new compound.
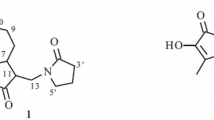
Compound 1 was obtained as a white amorphous powder, and the molecular formula, C21H28O7 was deduced from the positive-ion at m/z 415.2012 [M + Na]+ (calcd for C21H28O7Na, 415.2023) in the HR-ESI-MS spectrum. A strong absorption band at λmax 290 nm in the UV spectrum and an IR absorption band at 1762 cm–1 were indicative of the presence of a lactone moiety [10]. The 13C NMR spectrum (Table 1) displayed 15 carbon signals, including an ester carbonyl at δ 165.4 (C-12), an oxygenated olefinic quaternary carbon at δC 145.9 (C-6), and two olefinic quaternary carbons at δ 147.5 (C-7)/131.4 (C-11), indicating a γ-lactone group [10]. The NMR and HSQC spectra displayed signals for an olefinic group at δH 5.87 (s)/δC 125.4 (C-5), an oxygenated methylene [δH 4.64 (d, J = 13.2 Hz, H-13a) and 4.54 (d, J = 13.2 Hz, H-13b)/δC 54.5 (C-13)], three methylenes [δH 2.11 m/δC 38.0 (C-2), δH2.13 m/δC 39.5 (C-3), and δH 2.44 (dd, J = 15.6, 11.6 Hz, H-9′), 1.88 (m, H-9′)/δC 38.0 (C-9)], a methine at δH 1.92 (m)/δC 41.3 (C-10), a downfield shifted oxymethine at δH 6.32 (d, J = 8.0 Hz)/δC 70.1 (C-8), a ketal quaternary carbon at δC 109.0 (C-1), and an oxygenated quaternary carbon at δ 81.1 (C-4), indicative of a hirsutinolide-type sesquiterpene lactone [4]. In addition, the 1D and 2D NMR spectra revealed an olefinic signal at δH 6.17 (q, J = 7.6 Hz)/δC 139.9 (C-3′), two methyl groups downfield shifted [δH 2.04 (d, J = 7.6 Hz)/ δC 16.0 (C-4′) and δH 1.93 (s)/δC 20.5 (C-5′)], an olefinic quaternary carbon at δC 127.2 (C-2′), and an ester carbonyl carbon at δC 168.3 (C-1′), indicative of a tigloyl group possessing a Z configured olefinic (C-2′–C-3′) bond, which was quite similar to that of 8′-(2′Z-tigloyloxy)-hirsutinolide [10]. However, the 1H and 13C NMR spectra of 1 showed an additional signal for a methoxy group at δH 3.26 (s)/δC 49.5 (1-OCH3), indicating an attachment of this functional group at C-1 position.

This observation was further supported by the HMBC correlation between the methoxy proton (1-OCH3) and the ketal carbon (C-1). The relative stereochemistry of asymmetric carbons of 1 was deduced to be the same as that of 8 α-(2′Ztigloyloxy) hirsutinolide [10], based on its physicochemical data analyses. Thus, compound 1 was elucidated as a new compound, 8α-(2′Z-tigloyloxy)-1 α-methoxyhirsutinolide.
Compound 2 was isolated for the first time in nature and identified as 8α-(2′-hydroxymethylacryloyloxy)-1α-methoxyhirsutinolide-13-O-acetate by comparison of its physical and spectral data with published values [12].
Experimental
General Methods. Specific rotations were measured on a Rudolph Research Autopol IV multiwavelength polarimeter. UV spectra were run on a Shimadzu PharmaSpec-1700 UV/vis spectrophotometer. IR spectra were measured on a Bruker Tensor-27 FT-IR spectrometer. NMR spectroscopic data were recorded at room temperature on Bruker Avance DRX-400 MHz spectrometers, and the data were processed using Top Spin 3.1 software. High-resolution electrospray ionization mass spectra (HR-ESI-MS) were obtained with an Agilent 6530 LC-qTOF High Mass Accuracy mass spectrometer operated in the positive- and negative-ion modes. Analytical TLC was performed on 0.25 mm thick silica gel F254 glass-backed plates (Sorbent Technologies). Column chromatography was carried out with silica gel (230–400 mesh, Sorbent Technologies), Sephadex LH-20 gel (GE Healthcare), and RP-18 (YMC·GEL, 12 nm, S-150μm; Sorbent Technologies). Semipreparative (10 × 150 mm) columns were used for semipreparative HPLC, which was conducted on a Beckman Coulter Gold-168 system equipped with a photodiode array detector using an Alltech reversed-phase Econosil C18 column (10μm, 10 × 250 mm) with a flow rate of 1.5 mL/min.
Plant Material. The aerial parts of V. cinerea Less. (Asteraceae) were provided by Lampang Herb Conservation Club, Lampang Province, Thailand, in May 2011. The plant materials were identified by Dr. Thanapat Songsak (Faculty of Pharmacy, Rangsit University, Thailand). A voucher specimen (No. VCS02) has been deposited at the Natural Product Chemistry Laboratory, Daniel K. Inouye College of Pharmacy, University of Hawaii at Hilo.
Extraction and Isolation. The air-dried aerial parts of V. cinerea (1 kg) were extracted by maceration in MeOH (3 × 20 L) at room temperature. The solvent was concentrated in vacuo to yield 50 g of a crude extract, which was then suspended in distilled water (2 L) and then extracted successively with CHCl3 (3 × 2 L), EtOAc (3 × 2 L), and n-butanol (2 × 4 L). The CHCl3-soluble extract (20 g) was separated by column chromatography over Si gel (CC; ∅ 20 cm; 230–400 mesh, 2 kg) using a gradient solvent system of n-hexane–EtOAc (100:1 to 0:100) to afford 16 fractions (C1–C10). Combined fractions (30 g) from C3 to C7 were subjected to Si gel column chromatography (CC; ∅ 10 cm; 230–400 mesh, 500 g), with CHCl3–MeOH (100:0 to 1:1) as the solvent system, yielding 10 subfractions (C3S1–C3S10). Subfraction C3S5 (0.5 g) was rechromatographed on a Sephadex LH-20 gel column (CC; ∅ 5 cm; 200 g) to give two subfractions (C3S5L1–C3S5L2). The combined two subfractions were purified by HPLC on a semipreparative RP-18 column using MeOH–H2O mixtures (50:50 to 0:100) as the solvent system to yield 2 (1.5 mg, tR 110 min) and 1 (1.0 mg, tR 115 min).
8 α-(2′Z-Tigloyloxy)-1α -methoxyhirsutinolide (1). White amorphous powder; \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +30.0°(c 0.2, MeOH). UV (MeOH, λmax, nm) (log ε): 290 (4.0). IR (KBr, λmax, cm–1): 3320, 1762. 1H (400 MHz) and 13C NMR (100 MHz) data, see Table 1. HR-ESI-MS m/z 415.2012 [M + Na]+ (calcd for C21H28O7Na, 415.2023).
8α -(2′-Hydroxymethylacryloyloxy)-1α-methoxyhirsutinolide-13-O-acetate (2). White amorphous powder, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +25.0° (c 0.2, MeOH). UV (MeOH, λmax, nm) (log ε): 285 (3.90). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 6.42 (1H, d, J = 7.2, H-8), 6.34 (1H, s, H-3a′), 5.95 (1H, s, H-5), 5.77 (1H, s, H-3b′), 5.12, 5.07 (each 1H, d, J = 12.8, H-13a), 4.28 (2H, br.s, H-4′), 3.31 (3H, s, 1-OCH3), 2.12 (2H, m, H-2), 2.10 (2H, m, H-3), 1.85 (1H, m, H-10), 2.40 (1H, dd, J = 12.5, 12.5, H-9a), 1.91 (1H, ddd, J = 12.5, 7.6, 1.6, H-9b), 1.58 (3H, s, CH3-15), 0.94 (3H, d, J = 4.8, CH3-14). ESI-MS m/z 459 [M + Na]+.
References
N. J. Toyang and R. Verpoorte, J. Ethnopharmacol., 146, 681 (2013).
A. M. Abeysekera, K. T. D. De Silva, S. R. P. De Silva, V. D. P. Sirimanne, R. P. Labadie, A. J. J. Van den Berg, and W. Van der Sluis, Fitoterapia, 70, 317 (1999).
E. O. Iwalewa, J. O. Iwalewa, and J. O. Adeboye, J. Ethnopharmacol., 86, 229 (2003).
Y.-H. Kuo, Y.-J. Kuo, A.-S. Yu, M.-D. Wu, C.-W. Ong, L.-M. Y. Kuo, J.-T. Huang, C.-F. Chen, and S.-Y. Li, Chem. Pharm. Bull., 51, 425 (2003).
A. Chea, S. Hout, C. Long, L. Marcourt, R. Faure, N. Azas, and R. Elias, Chem. Pharm. Bull., 54, 1437 (2006).
C. Gunasingh, G. Barnabas, and S. Nagarajan, Indian J. Pharm. Sci., 43, 114 (1981).
T. N. Misra, R. S. Singh, J. Upadhyay, and R. Srivastava, Phytochemistry, 23, 415 (1984).
P. Pratheeshkumar and G. Kuttan, Immunopharmacol. Immunotoxicol., 33, 533 (2011).
M. Tandon, Y. N. Shukla, A. K. Tripathi, and S. C. Singh, Phytother. Res., 12, 195 (1998).
U. J. Youn, G. Miklossy, X. Chai, S. Wongwiwatthananukit, O. Toyama, T. Songsak, J. Turkson, and L. C. Chang, Fitoterapia, 93, 194 (2014).
G. Miklossy, U. J. Youn, P. Yue, M. Zhang, C.-H. Chen, T. S. Hilliard, D. Paladino, Y. Li, J. Choi, J. N. Sarkaria, J. K. Kawakami, S. Wongwiwatthananukit, Y. Chen, D. Sun, L. C. Chang, and J. Turkson, J. Med. Chem., 58, 7734 (2015).
F. Bohlmann, G. Brindoepke, and R. C. Rastogi, Phytochemistry, 17, 475 (1978).
Acknowledgment
This research was supported by the National Institute of Minority Health and Health Disparities (NIMHD) of the National Institutes of Health under award number U54MD008149 (L.C.C). The content is solely the responsibility of the authors and does not necessarily represent the official view of NIMHD or NIH. This work was also supported by the Korea Polar Research Institute, KOPRI, under the project PE17100 (U.J.Y). We thank H. S. Shin, National Center for Inter-university Research Facilities, Seoul National University, for providing the mass spectrometry facility used in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2018, pp. 196–198.
Rights and permissions
About this article

Cite this article
Youn, U.J., Wongwiwatthananukit, S., Songsak, T. et al. Sesquiterpene Lactones from Vernonia cinerea. Chem Nat Compd 54, 235–237 (2018). https://doi.org/10.1007/s10600-018-2311-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-018-2311-5