
One new phthalate, named kurraminate (1), has been isolated from Nepeta kurramensis Rech. f., along with five known compounds, bis(2-ethylicosyl)phthalate (2), ursolic acid (3), β-sitosterol (4), β-amyrin (5), and 1-nonacosanol (6). The structure of the compounds was identified on the basis of their 1D and 2D NMR and mass spectrometry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The genus Nepeta belongs to the family Labiatae (Lamiaceae), and this genus is one of the largest genera of Labiatae [1]. Plants belonging to Nepeta are used in traditional medicine, viz. as vulnerary, diuretic, diaphoretic, antiasthmatic, antispasmodic, febrifuge, tonic, and sedative agents [2,3,4]. It is interesting to note that some Nepeta species are used for the treatment of gastrointestinal diseases and different disorders, viz. respiratory and nervous disorders [4]. Various compounds and ingredients reported earlier from the genus Nepeta control calcineurin in vitro [5, 6]. Some members of the genus are also used as an insect repellent in various countries of the world [7, 8]. The medicinal properties of these compounds reported earlier from the different species of the genus prompted us to carry out phytochemical investigation on the present species, Nepeta kurramensis, which has rarely been explored phytochemically. In this paper we present the isolation and structural elucidation of one new and five known compounds which, to the best of our knowledge, are isolated for the first time from Nepeta kurramensis.
The molecular formula of compound 1 was established as C48H82O6 (eight degrees of unsaturation) through HR-EI-MS analysis, which gave a molecular ion peak at m/z 754.6107 (calcd for C48H82O6, 754.6111). This was supported by 1H, 13C, and DEPT NMR spectroscopic data. Further fragments were observed in the EI-MS, which indicated the presence of the phthalate moiety and length of side chain, as supported by NMR spectral data. The IR spectrum of 1 exhibited absorption bands at 3345 (OH), 2814 (C–H stretch), 1725 (C=O), 1611 (aromatic), 1575, 1445, and 745 cm–1 in the molecule. The UV maxima showed the presence of a conjugated system at 276 nm.

In the 1H NMR spectrum of 1, two important signals at δ 7.51 (2H, m, H-2, 5) and 7.69 (2H, m, H-3, 4) (see Experimental) indicated the presence of the aromatic region in the molecule. The chemical shift values and the splitting pattern were characteristic of a 1,2-disubstituted benzene ring. The downfield signal at δ 4.27 (4H, m, H-3′, 3″) indicated the presence of two sets of oxygenated methylene groups. The 1H NMR spectrum also showed a methine signal at δ 2.30 (2H, m, H-4′, 4″), a methylene signal at δ 3.71 (4H, br.d, J = 7.0 Hz, H-22′, 22″), and a methyl signal at δ 0.91 (6H, t, J = 7.5 Hz, H-21′, 21″). All these signals in the 1H NMR spectrum coupled with the 13C NMR spectral data indicated the phthalate nature of the molecule [9,10,11,12,13,14,15], with two identical side chains. Four degrees of unsaturation was satisfied by the benzene ring, whereas the presence of two olefinic moieties and two carbonyl groups further supported the phthalate skeleton [9,10,11,12,13,14,15]. The olefinic moieties in the molecule were indicated by the presence of signals at δ 5.32 (2H, m, H-5′, 5″) and 5.24 (2H, m, H-6′, 6″), whereas the long chains of methylenes were confirmed by the multiplets at δ 1.23–1.27 (m, H-8′–15′ and H-8″–15″).
Analysis of the 13C NMR spectrum and the DEPT experiments showed the presence of one methyl group, 16 methylene carbons (two O-bearing methylenes), two sp2 aromatic carbons, three methines, and one quaternary and one carbonyl carbon for half of the molecule. The resonances at δ 173.2, 132.4, 130.8, 129.8, and 62.07 indicated the presence of the phthalate moiety, while the signals at δ 68.1 were assigned to the C-22″/22″ oxymethylene groups in the molecule. The signals observed at δ 38.7, 34.2, 32.5, 30.3, 29.7–27.2, 24.8, 23.7, 22.9, and 22.6 indicated the presence of methylene carbons with double intensity, which was half of the number observed in the MS spectrum (Fig. 1), indicating two identical parts in 1.

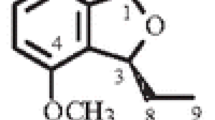
a: Mass fragmentation pattern in compound 1; b: key COSY and HMBC interactions for compound 1.
Analysis of the 1H and 13C NMR spectral data and the coupling constants for four aromatic protons led to the identification of the ortho-substituted aromatic ring. The multiplicities of carbons were determined by DEPT experiment, and the complete assignments were made on the basis of COSY, HMQC, and HMBC techniques (Fig. 1). The length of the side chain and the position of the ethyl groups were established through mass fragmentation patterns, as illustrated in Fig. 1. The position of side chain was established on the basis of HMBC correlations of H-3′/3″ to C-1′/C-1″; H-2/5 to C-1/6, 4/3, and C-1′/1″; H-22′/22″ to C-3′/3″, C-5′/5″, and C-6′/6″; H-5′/5″to C-4′/4″ and C-6′/6″; and H-3′/3″ to C-1′/1″. Consequently, the structure of compound 1 was established as bis(E)-2-(hydroxymethyl)nonadec-3-enyl)phthalate, named kurraminate, after the producing organism, Nepeta kurramensis.
Structure elucidation of the five known compounds, viz. bis(2-ethylicosyl)phthalate (2) [11, 12], ursolic acid (3) [16], β-sitosterol (4) [17], β-amyrin (5) [18], and 1-nonacosanol (6) [19], was achieved on the basis of the NMR and MS spectral data and their comparison with those reported in the literature.
Experimental
General Experimental Procedures. The UV (MeOH) spectra were recorded in methanol on a Hitachi U-3200 spectrometer (λmax in nm). The IR (KBr) spectra were measured on Shimadzu-8900 spectrophotometer (v in cm–1). EI-MS and HR-EI-MS were carried out using MAT 8200 and Micromass LCT mass spectrometers, in m/z. The 1H NMR spectrum was recorded on Bruker AMX-500 instrument using TMS as an internal reference. The chemical shifts are recorded in ppm (δ), and the coupling constants (J) in Hertz. The 13C NMR spectra were recorded at 125 MHz on the same instrument.
Plant Material. The plant, Nepeta kurramensis, was collected from Kurram Agency, Khyber Pakhtunkhwa, Pakistan in July 2009 and identified by the plant taxonomist at the Department of Plant Sciences, Kohat University of Science and Technology, Kohat. A voucher specimen of this plant (PLS/Herb 1016) is deposited in the Herbarium of the Kohat University for future reference. After collection, the selected species was washed under running tap water, air dried, and homogenized to a fine powder.
Extraction and Isolation. The air-dried whole powder plant material of N. kurramensis (7.5 kg) was soaked in methanol (10 L) for three weeks and filtered. The filtrate was evaporated under reduced pressure using a rotary evaporator to get a crude greenish extract, which was extracted with n-hexane, CHCl3, EtOAc, n-butanol, and water. The chloroform extract (65 g) was subjected to column chromatography on silica gel using n-hexane, n-hexane–CHCl3, CHCl3–MeOH and finally with pure MeOH as mobile phases, which yielded seven fractions (NS1–NS7).
Fraction NS3 of the first column was loaded on silica gel and eluted with CHCl3–n-hexane (7.5:2.5) to give kurraminate (1, 11.7 mg). Compound 2 (25.5 mg) was purified from the same fraction by eluting with CHCl3–n-hexane (5.5:4.5). The main fraction NS4 (5.2 g) obtained from the first column (60% chloroform–n-hexane) showed two spots on TLC with some impurities and thus was further purified by repeated silica gel column chromatography to obtain ursolic acid (3, 14.3 mg) and β-sitosterol (4, 12.4 mg). Fraction NS6 of the first column showed one spot on TLC with some impurities and was further purified by repeated silica gel CC and eluted with CHCl3–n-hexane (6:4), yielding β-amyrin (5, 16.5 mg). Similarly, 1-nonacosanol (6, 12.0 mg) was obtained from fraction NS2 after elution with CHCl3–n-hexane (3:7).
Kurraminate (1). Yellowish oil. UV (CH2Cl2, λmax, nm): 276. IR (KBr, cm–1): 3345 (OH), 2814 (C–H), 1725 (C=O), 1611, 1575, 1445, 745 (aromatic). 1H NMR (500 MHz, CDCl3, δ, ppm, J/Hz): 7.51 (2H, m, H-2, 5), 7.69 (2H, m, H-3, 4), 5.32 (2H, m, H-5′, 5″), 5.24 (2H, m, H-6′, 6″), 4.27 (4H, m, H-3′, 3″), 3.71 (4H, br.d, J = 7.0, H-22′, 22″), 2.30 (2H, m, H-4′, 4″), 1.58 (2H, br.s, H-7′, 7″), 1.43 (8H, m, H-8′, 8″), 1.23–1.27 (remaining CH2), 0.91 (6H, t, J = 7.5, H-21′, 21″). 13C NMR (125 MHz, CDCl3,δ, ppm): 173.2 (C-1′, 1″), 132.4 (C-1, 6), 130.8 (C-3, 4), 129.8 (C-2, 5), 128.7 (C-5′, 5″), 130.0 (C-6′, 6″), 68.1 (C-22′, 22″), 62.0 (C-3′, 3″), 38.7 (C-4′, 4″), 34.2 (C-7′, C-7″), 32.5 (C-8′, 8″), 30.3 (C-9′, 9″), 29.7–27.2 (remaining CH2), 24.8 (C-17′, 17″), 23.7 (C-18′, 18″), 22.9 (C-19′, 19″), 22.6 (C-20′, 20″), 14.1 (C-21′, 21″). HR-EI-MS m/z 754.6107 [M]+ (calcd for C48H82O6, 754.6111).
References
J. Hussain, N. U. Rehman, H. Hussain, A. Al-Harrasi, L. Ali, T. S. Rizvi, M. Ahmad, and Mehjabeen, Fitoterapia, 83, 593 (2012).
T. Dirmenci, B. Yildiz, and G. Tumen, Turk. J. Bot., 28, 221 (2004).
N. Miceli, M. F. Taviano, D. Giuffrida, A. Trovato, O. Tzakou, and E. M. Galati, J. Ethnopharmacol., 97, 261 (2005).
D. S. Bisht, R. C. Padalia, L. Singh, V. Pande, P. Lal, and C. S. Mathela, J. Serb. Chem. Soc., 75, 739 (2010).
Y. Cigremis, Z. Ulukanli, A. Ilcim, and M. Akgoz, Eur. Rev. Med. Pharmacol. Sci., 14, 661 (2010).
T. A. K. Prescott, N. C. Veitch, and M. S. J. Simmonds, J. Ethnopharmacol., 137, 1306 (2011).
J. J. Zhu, X. P. Zeng, D. Berkebile, H. J. Du, Y. Tong, and K. Qian, Med. Vet. Entomol., 23, 209 (2009).
M. A. Birkett, A. Hassanali, S. Hoglund, J. Pettersson, and J. A. Pickett, Phytochemistry, 72, 109 (2011).
J. Hussain, N. U. Rehman, H. Hussain, L. Ali, A. Al-Harrasi, and V. U. Ahmad, Helv. Chim. Acta, 94, 2106 (2011).
J. Hussain, N. Rehman, H. Hussain, L. Ali, and A. Al-Harrasi, J. Asian Nat. Prod. Res., 14, 22 (2012).
K. S. Satyan, A. Prakash, R. P. Singh, and R. S. Srivastava, Planta Med., 61, 293 (1995).
M. Saleem, M. Nazir, N. Akhtar, A. P. Onochab, N. Riaza, A. Jabbar, S. M. Ali, and N. Sultana, J. Asian Nat. Prod. Res., 11, 974 (2009).
Y. Li, Z. J. Qian, and S. K. Kim, Bioorg. Med. Chem. Lett., 18, 6130 (2008).
M. A. A. Al-Bari, M. A. Sayeed, M. S. Rahman, and M. A. Mossadik, Res. J. Med. Med. Sci., 1, 77 (2006).
S. J. Uddin, J. Bettadapura, P. Guillon, D. Grice, S. Mahalingam, and E. Tiralongo, J. Antivir. Antiretrovir., 5, 139 (2013).
J. Hussain, F. Ullah, H. S. Hussain, and S. T. Hussain, Z. Naturforsch., 63b, 591 (2008).
M. Hasan, D. K. Burdi, and V. U. Ahmad, Phytochemistry, 30, 4181 (1991).
M. Saleem, A. Farooq, S. Ahmad, N. Shafiq, N. Riaz, A. Jabbar, M. Arshad, and A. Malik, J. Asian Nat. Prod. Res., 12, 702 (2010).
H. R. El-Seedi, T. Ringbom, K. Torssell, and L. Bohlin, Chem. Pharm. Bull., 51, 1439 (2003).
Acknowledgment
The authors wish to thank the Higher Education Commission (HEC), Government of Pakistan, for providing financial support for the current study under the National Research Program for Universities (NRPU).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2017, pp. 363–365.
Rights and permissions
About this article

Cite this article
Rehman, N.U., Ahmad, N., Hussain, J. et al. One New Phthalate Derivative from Nepeta kurramensis . Chem Nat Compd 53, 426–428 (2017). https://doi.org/10.1007/s10600-017-2014-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-017-2014-3