
A new isochroman analogue, named monascuspilorin (1), was obtained from the n-BuOH-soluble fraction of the 95% EtOH extract of red mold rice fermented with the fungus Monascus pilosus BCRC 38072 (Eurotiaceae). The structure of the new compound was elucidated by comprehensive spectroscopic analysis, especially HR-ESI-MS and NMR experiments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The genus Monascus (family Monascaceae, class Ascomycetes) comprises five representative species: M. pilosus, M. purpureus, M. ruber, M. kaoliang, and M. anka [1, 2]. Red yeast rice, which is also known as red fermented rice and red mold rice, is produced by growing Monascus sp. on rice to produce a red-colored product. Red yeast rice is obtained by the fermentation of rice with fungi of the genus Monascus, mainly M. pilosus, M. purpureus, and M. anka, to produce a red-colored product [1]. Red fermented rice has been used as a traditional food additive for improving the color of meat, fish, and soybean products, and is also described to have conserving properties in oriental countries for centuries. Fungi of the genus Monascus are known to produce several secondary metabolites, including (I) a group of antihypercholesterolemic agents that include monacolin K and a hypotensive agent, γ-aminobutyric acid (GABA) [3], (II) a group of pigments (orange pigments, monascorubrin and rubropunctanin; yellow pigments, ankaflavin and monascin; and red pigments, monascorubramine and rubropuctamine) [4], (III) antioxidant compounds including dimerumic acid [5] and 3-hydoxy-4-methoxybenzoic acid [6], and (IV) antibacterial compounds including pigments and citrinin [4, 7]. Recently, there have also been many reports on these secondary metabolites [8,9,10,11,12,13,14,15,16,17,18,19]. Careful examination on the n-BuOH-soluble fraction of a 95% EtOH extract of the red mold rice produced by Monascus pilosus led to the isolation of a new isochroman analog, monascuspilorin (1). We herein reported on the isolation and structure elucidation of the new metabolite.
Compound 1 was obtained as an optically active oil with [α] 22D –115.8° (c 0.075, MeOH). The molecular formula was determined as C25H40O7 on the basis of the [M + Na]+ peak at m/z 475.2672 (calcd for C25H40NaO7, 475.2677) in its HR-ESI-MS. The UV absorption (λmax 275 nm) and the exhibition of a bathochromic shift in alkaline solution suggested the presence of a phenolic isochroman skeleton [20]. The bands at 3430, 1585, and 1470 cm–1 in the IR spectrum revealed the presence of OH groups and the benzene ring. Six indices of hydrogen deficiency (IHD) were determined from the molecular formula, 13C NMR (Table 1), and DEPT spectra.
The 1H NMR/13C NMR spectra of 1 (Table 1) showed signals of an n-octyl moiety [δH 0.92 (3H, t, J = 7.5 Hz, CH3-16), 1.38–1.42 (10H, br.s, CH2-11–15), 1.48/1.62 (each 1H, m, H-10b, 10a), 1.65 (2H, m, CH2-9); δC 14.0 (C-16), 23.7 (C-15), 33.1 (C-14), 29.6 (C-13), 29.7 (C-12), 29.5 (C-11), 25.8 (C-10), and 37.2 (C-9)]. Signals of two Me groups attached to aromatic rings were detected at δH 2.04 (3H, s, CH3-17) and 2.17 (3H, s, CH3-18), and this finding was further supported by two peaks in the 13C NMR spectrum, δC 12.0 (C-17) and 10.4 (C-18). One nonequivalent methylene proton at δH 2.40 (1H, dd, J = 16.1, 12.0 Hz, H-3β), 2.65 (1H, dd, J = 16.1, 3.0 Hz, H-3α), and δC 33.5 (C-3), an oxymethine at δH 3.58 (1H, m, H-2) and δC 76.5 (C-2), and one nonequivalent oxymethylene at δH 4.60/4.95 (each 1H, d, J = 14.0 Hz, CH2-8) and δC 66.7 (C-8) were also observed.
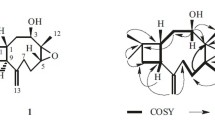
In addition, individual sugar protons (rhamnosyl unit) were deduced using COSY and HSQC experiments [δH 1.33 (3H, d, J = 6.5 Hz, CH3-6′), 3.44 (1H, dd, J = 9.6 Hz, H-4′), 3.82 (1H, dd, J = 9.6, 3.6 Hz, H-3′), 4.04 (1H, dq, J = 9.6, 6.5 Hz, H-5′), 4.18 (1H, dd, J = 3.6, 2.2 Hz, H-2′), 4.81 (1H, d, J = 2.2 Hz, H-1′); δC 18.0 (C-6′), 71.6 (C-5′), 72.4 (C-4′), 73.2 (C-3′), 72.0 (C-2′), 107.2 (C-1′)]. The glycosidic bond was α-oriented according to the small coupling constant (J = 2.2 Hz) of the anomeric proton at δ4.81. The 1H, 13C NMR (Table 1), and DEPT spectra exhibited signals for four primary carbons δ10.4 (C-18), 12.0 (C-17), 14.0 (C-16), and 18.0 (C-6′), nine secondary carbons including one O-bearing, δ 66.7 (C-8), 37.2 (C-9), 33.5 (C-3), 29.5 (C-11), 25.8 (C-10), 29.7 (C-12), 29.6 (C-13), 33.1 (C-14), and 23.7 (C-15), six tertiary methines including one hemiacetal δ 107.2 (C-1′), 76.5 (C-2), 72.0 (C-2′), 72.4 (C-4′), 73.2 (C-3′), and 71.6 (C-5′), and six quaternary carbons δ 153.2 (C-5), 154.2 (C-7), 131.7 (C-3a), 120.6 (C-7a), 121.0 (C-4), and 117.5 (C-6). The 1H, 13C NMR, and DEPT spectra indicated that the structure of 1 has a fully substituted benzene ring. The COSY spectra (Fig. 1) showed the contacts between the fragments of n-octyl and the rhamnosyl moeity of 1.

The structure and key COSY and HMBC correlations of 1.
Further confirmation by the HMBC correlations (Fig. 1) of CH2-3/C-2, C-4, C-9, C-3a, C-7a, and CH2-8/C-2, C-7, C-3a, and C-7a, verified the main skeleton of 1. The location of the n-octyl moiety attached at C-2 was determined by the HMBC experiment, in which cross-peaks were observed between CH2-3/C-9 and H-2/C-10. The α-orientation O-linkage of the rhamnosyl part and the main skeleton via C-7 was demonstrated by the corresponding HMBC correlations of H-1′/C-7, as shown in Fig. 1. By further analyses of the HSQC, HMBC, and COSY spectra, the proton and carbon signals were assigned unambiguously, which resulted in the establishment of the planar structure for compound 1.
The relative configuration of compound 1 was established via ROESY data and Chem3D modeling. The α-orientation of the n-octyl unit at C-2 was deduced from the observation of the quasi-1,3-diaxial interactions between Hβ-2 and Hβ-8. From the above data, compound 1 was unambiguously characterized as rel-(2S)-4,6-dimethyl-2-octyl-7-O-α-L-rhamnosylisochroman-5-ol, named monascuspilorin, and its structure was established as 1, which was further confirmed by COSY and HMBC (Fig. 1).
Experimental
General Experimental Procedures. Optical rotations were measured on a Jasco P-1020 digital polarimeter, UV spectra were obtained on a Jasco UV-240 spectrophotometer in MeOH, and IR spectra (KBr or neat) were taken on a PerkinElmer System 2000 FT-IR spectrometer. 1D (1H, 13C, DEPT) and 2D (COSY, ROESY, HSQC, HMBC) NMR spectra using CDCl3 as solvent were recorded on Varian VNMRS 600 (600 MHz for 1H NMR, 150 MHz for 13C NMR) spectrometers. Chemical shifts were internally referenced to the solvent signals in methanol-d4 (1H, δ 3.31; 13C, δ 49.0) with TMS as the internal standard. Low-resolution ESI-MS spectra were obtained on an API 3000 (Applied Biosystems), and high-resolution ESI-MS spectra on a Bruker Daltonics APEX II 30e spectrometer. Low-resolution EI-MS spectra were recorded on a Quattro GC/MS spectrometer having a direct inlet system. Silica gel (70–230, 230–400 mesh) (Merck) was used for column chromatography, and silica gel 60 F-254 (Merck) was used for TLC and preparative TLC.
Fungus Material. Monascus pilosus BCRC 38072 was used throughout this study, and specimens were deposited at the Bioresource Collection and Research Center (BCRC) of the Food Industry Research and Development Institute (FIRDI). M. pilosus was maintained on potato dextrose agar (PDA, Difco). The strain was cultured on PDA slants at 25°C for 7 days, and the spores were harvested by sterile water. The spores (5 × 105) were seeded into 300 mL shake flasks containing 50 mL RGY medium (3% rice starch, 7% glycerol, 1.1% polypeptone, 3.2% soybean powder, 0.2% MgSO4, 0.2% NaNO3) and cultivated with shaking (150 rpm) at 25°C for 3 days. After the mycelium enrichment step, an inoculum mixing 100 mL mycelium broth and 100 mL RGY medium was inoculated into plastic boxes (25 cm 30 cm) containing 1 kg sterile rice and cultivated at 25°C for producing red mold rice (RMR; also called beni-koji in Japan). At day 7, 150 mL RGY medium was added to maintain the growth of cells. After 14 days of cultivation, the RMR was harvested and lyophilized for the extraction of metabolites.
Extraction and Separation of Compounds. The dried red mold rice of Monascus pilosus BCRC 38072 (1.0 kg) was extracted five times with 95% EtOH at room temperature. The ethanolic syrup extract was partitioned between n-BuOH–H2O (1:1) to afford n-BuOH (1.8 g) and H2O soluble fractions. The n-BuOH-soluble fraction (Fr. A, 1.8 g) was chromatographed by CC (60 g SiO2, 70–230 mesh), eluting with acetone and enriched with MeOH to produce 10 fractions: Fr. 1–10. Fraction 5 was subjected to RP-18 silica gel CC using H2O–acetone (1:1) as the eluent to obtain eight fractions: Fr. 5.1–5.8. Fraction 5.5 was subjected to RP-18 preparative TLC with CH2Cl2–MeOH (30:1) to yield 1 (2.5 mg).
Monascuspilorin (1). Yellow oil, [α] 22D –115.8° (c 0.075, MeOH). IR spectrum (neat, νmax, cm–1): 3430 (OH), 1585, 1470 (benzene ring). UV (MeOH, λ, nm) (lg ε): 275 (4.65). 1H and 13C NMR, see Table 1. ESI-MS 475 [M + Na]+. HR-ESI-MS 475.2672 [M + Na]+ (calcd for C25H40NaO7 +, 475.2677).
References
R. E. Mudgett, Monascus, in: Natural Food Colorants: Science and Technology, G. J. Lauro, F. J. Francis (eds.), Marcel Dekker, New York, 2000, p. 31.
J. Ma, Y. Li, Q. Ye, J. Li, Y. Hua, D. Ju, D. Zhang, R. Cooper, and M. Chang, J. Agric. Food Chem., 48, 5220 (2000).
Y. C. Su, J. J. Wang, T. T. Lin, and T. M. Pan, J. Ind. Microbiol. Biotechnol., 30, 40 (2003).
H. C. Wong and P. E. Koehler, J. Food Sci., 46, 589 (1981).
Y. Aniya, I. I. Ohtani, T. Higa, C. Miyagi, H. Gibo, M. Shimabukuro, H. Nakanish, and J. Taira, Free Radic. Biol. Med., 286, 999 (1999).
G. F. Wu and X. C. Wu, Acta Microbiol. Sin., 40, 394 (2000).
P. J. Blanc, M. O. Loret, and G. Goma, Biotechnol. Lett., 17, 291 (1995).
Ming-Der Wu, Ming-Jen Cheng, Tai-Wei Liu, Yen-Lin Chen, Hing-Yuen Chan, Hui-Ping Chen, Wen-Jung Wu, Kai-Ping Chen, and Gwo-Fang Yuan, Chem. Nat. Compd., 51, 554 (2015).
K. Tsuji, T. Ichikawa, N. Tanabe, S. Abe, S. Tarui, and Y. Nakagawa, Nippon. Nogeikagaku Kaishi., 66, 1241 (1992).
A. Endo, J. Med. Chem., 28, 401 (1985).
T. Akihisa, H. Tokuda, K. Yasukawa, M. Ukiya, A. Kiyota, N. Sakamoto, T. Suzuki, N. Tanabe, and H. Nishino, J. Agric. Food Chem., 53, 562 (2005).
M. J. Cheng, M. D. Wu, I. S. Chen, and G. F. Yuan, Chem. Pharm. Bull., 56, 394 (2008).
S. Jongrungruangchok, P. Kittakoop, B. Yongsmith, R. Bavovada, S. Tanasupawat, N. Lartpornmatulee, and Y. Thebtaranonth, Phytochemistry, 65, 2569 (2004).
Z. Huang, Y. Xu, L. Li, and Y. Li, J. Agric. Food Chem., 56, 112 (2008).
P. Juzlova, T. Rezanka, L. Martinkova, and V. Kren, Phytochemistry, 43, 151 (1996).
L. L. Li, J. P. Chen, and L. Y. Kong, Chin. J. Nat. Med., 4, 32 (2006).
D. Wild, G. Toth, and H. U. Humpf, J. Agric. Food Chem., 51, 5493 (2003).
K. Sato, Y. Goda, S. S. Sakamoto, H. Shibata, T. Maitani, and T. Yamada, Chem. Pharm. Bull., 45, 227 (1997).
M. O. Loret and S. Morel, J. Agric. Food Chem., 58, 1800 (2010).
W. A. Ayer and S. Miao, Can. J. Chem., 71, 487 (1993).
Acknowledgment
This investigation was supported by a grant from the Ministry of Economic Affairs of the Republic of China.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2017, pp. 39–41.
Rights and permissions
About this article

Cite this article
Cheng, MJ., Wu, MD., Chan, HY. et al. New Metabolite Isolated from the Fungus Monascus pilosus . Chem Nat Compd 53, 44–47 (2017). https://doi.org/10.1007/s10600-017-1907-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-017-1907-5