
Chemical investigation on the endophytic fungus Penicillium sp. B21 from the leaf of Bruguiera sexangula var. rhynchopetala led to the isolation of a new compound, penicitrinone acetate (1), the structure of which was elucidated by spectroscopic methods, including 2D NMR techniques.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In recent years, secondary metabolites obtained from plant or marine-derived fungi have gained considerable attention, as many of them are structurally unique and of great potential for medicinal and agricultural applications [1–4]. Although endophytic fungi asymptomatically invade host tissues, their secondary metabolites can stimulate plant growth or provide defense against plant pathogen attacks [5]. These microorganisms have thus been recognized as potential sources of new bioactive compounds [6]. Scientists have investigated the chemistry of mangrove plants since 1913, and mangrove plants have proved to be a well-established source for structurally diverse and biologically active secondary metabolites. More than 200 species of endophytic fungi have been isolated and identified from mangrove plants, and several reports on the isolation of antibiotic compounds from endophytic fungi have been reported [7–10].
In a systemic investigation, a new penicitrinone derivative, penicitrinone acetate (1), was isolated from the fermentation broth by endophytic fungus Penicillium sp. B21 in the leaf of Bruguiera sexangula. In this paper, we describe the isolation and structure of the new compound.
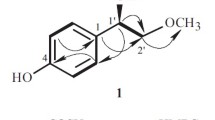
Compound 1, a red powder, had the molecular formula C25H26O6 on the basis of HR-ESI-MS m/z 423.1802 [M + H]+ (calcd for C25H27O6, 423.1808). The 1H NMR spectrum (Table 1) showed four secondary Me groups (δ 1.33, 1.34, 1.36, 1.46; 4d) and three tertiary Me groups (δ 1.96, 2.05, 2.19; 3s). As determined by 13C NMR spectrum (Table 1), the downfield signals corresponded to two CO groups (δ 172.2 and 184.2), six olefinic C-atoms (δ 100.3, 102.6, 131.7, 133.1, 157.3, and 159.0), six aromatic C-atoms (δ 103.3, 117.6, 136.5, 138.7, 140.5, and 148.5), and two O-bearing C-atoms (δ 83.2 and 88.5); the shielded region showed seven Me (δ 10.6, 11.7, 18.7, 19.0, 19.4, 20.6, and 21.1) and two CH groups (δ 35.6, 45.3). The NMR spectra of 1 were very similar to those of penicitrinone A [11], suggesting that 1 has the same skeleton as penicitrinone A. Comparison of the NMR spectra of 1 and penicitrinone A showed that the OH group at C-5′ in penicitrinone A was replaced by an AcO group in 1, which was supported by the NMR signal at δ 1.96 (3H, s) and δC 172.2 and 21.1, as well as the HMBC correlation of δ 1.96 (3H, s, 9-Me) with δ 172.2 (C-9). Finally, the structure of 1 was unambiguously established by detailed analysis of HSQC, HMBC, and ROESY spectrum (Fig. 1).

Key HMBC and ROESY correlations of compound 1.
Experimental
General Experimetal Procedures. Optical rotations were run on a JASCO P-1020 polarimeter at room temperature. IR spectra were recorded on an Avatar 360 FT-IR ESP spectrometer. Mass spectra were determined on a Bruker Apex 7.0 TESLA FT-MS apparatus for HR-ESI-MS. 1H and 13C NMR spectra were taken on a Bruker Av-400 spectrometer in acetone-d6. Analytical TLC was run on silica gel plates (GF254, Yantai Institute of Chemical Technology, Yantai, China). Spots on the plates were observed under UV light and visualized by spraying with 5% H2SO4 in ethanol followed by heating. Column chromatography (CC) was performed on silica gel (200–300 mesh and 300–400 mesh; Qingdao Marine Chemical Factory, Qingdao, China).
Isolation of the Metabolites from Penicillium sp. B21. The endophytic fungus Penicillium sp. B21 was separated from the leaf of Bruguiera sexangula var. rhynchopetala in Dong Zhai Gang, Hainan Province, China. Penicillium sp. B21 was cultured on moist rice (1200 g) in 40 conical flask at 28°C for 40 days. The fermented rice was extracted with 95% EtOH at room temperature, and the organic layer was evaporated in vacuo. The residue was suspended in H2O and extracted with EtOAc, and the organic layer was evaporated in vacuo to give a residue (116 g). The residue (100 g) was subjected to CC on silica gel (200–300 mesh, 2 kg, 10 × 120 cm) and eluted successively with petroleum ether (PE)–EtOAc (10:1, 5:1, 3:1, 1:1, 1:3, v/v) and EtOAc to yield fractions A–F. Fraction D (20 g) was subjected to repeated silica gel CC with chloroform–methanol (40:1–1:1, v/v) to give seven subfractions, and the third fractions was purified by recrystallization in PE–acetone (10:1, v/v) to afford a brown powder. The powder was purified further by Sephadex LH-20 gel column chromatography with chloroform–methanol (1:1, v/v) as eluant to afford 1 (550 mg).
Compound 1. Red amorphous powder. [α] 25D +2.2° (c 0.1, MeOH). IR (KBr, cm–1): 2969, 2927, 1615, 1510, 1390, 1336, 1285, 1193,1134, 895, 840. HR-ESI-MS m/z 423.1802 [M + H]+ (calcd for C25H27O6, 423.1808). Its 1H and 13C NMR signal assignments are summarized in Table 1.
References
M. E. Rateb and R. Ebel, Nat. Prod. Rep., 28, 290 (2011).
A. H. Aly, A. Debbab, J. Kjer, and P. Proksch, Fungal Divers., 41, 1 (2010).
D. Ronsberg, A. Debbab, A. Mandi, V. Wray, H. F. Dai, T. Kurtan, P. Proksch, and A. H. Aly, Tetrahedron Lett., 54, 3256 (2013).
A. Debbab, A. H. Aly, and P. Proksch, Fungal Divers, 49, 1 (2011).
J. M. Gao, S. X. Yang, and J. C. Qin, Chem. Rev., 113, 4755 (2013).
J. Xian, Q. Zhang, and Y. Q. Gao, J. Agric. Food Chem., 62, 3584 (2014).
J. Wu, Q. Xiao, J. Xu, M. Y. Li, J. Y. Pan, and M. H. Yang, Nat. Prod. Rep., 25, 955 (2008).
O. F. Smetanina, A. N. Yurchenko, M. V. Pivkin, E. A. Yurchenko, and Sh. Sh. Afiyatullov, Chem. Nat. Compd., 47, 118 (2011).
X. C. Liu, H. Li, F. Zhou, and R. M. Wang, Chem. Nat. Compd., 51, 1199 (2015).
A. N. Yurchenko, O. F. Smetanina, N. N. Kirichuk, E. A. Yurchenko, and Sh. Sh. Afiyatullov, Chem. Nat. Compd., 49, 1123 (2014).
D. Wakana, T. Hosoe, T. Itabashi, K. Okada, G. M. Campos Takaki, T. Yaguchi, K. Fukushima, and K. I. Kawai, J. Nat. Med., 60, 279 (2006).
Acknowledgment
This work was financially supported by Natural Science Foundation of Hainan Province, P. R. China (No. 214023) and Key Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education, P. R. China.
Author information
Authors and Affiliations
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2016, pp. 696–697.
Rights and permissions
About this article

Cite this article
Deng, PF., Luo, YP., Niu, YY. et al. A New Penicitrinone Derivative from the Endophytic Fungus Penicillium sp. from Bruguiera sexangula var. rhynchopetala . Chem Nat Compd 52, 810–812 (2016). https://doi.org/10.1007/s10600-016-1784-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-016-1784-3