
A phytochemical investigation of the roots and stems of Rubus alceaefolius Poir. let to the isolation of two oleanane-type triterpenoids, 2α,3α-dihydroxyolean-11,13(18)-dien-19β,28-olide ( 1 ) and 2α,3α-dihydroxyolean-13(18)-en-28-oic acid ( 2 ), and a unique octanordammarane triterpenoid, 3β-hydroxy- 20,21,22,23,24,25,26,27-octanordammaran-17β-ol ( 3 ), which the current study is the second report of this type of dammarane triterpenoid. Their structures were elucidated on the basis of spectroscopic evidence, including 1D and 2D NMR and IR analysis, and by comparison with literature data. Compounds 1 and 3 are new compounds, and compound 2 is obtained from nature for the first time.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Rubus alceaefolius Poir., one of the plants of the Rubus L. (Rosaceae) genus, is a Chinese herb used in the treatment of nasopharyngeal carcinoma, hepatic carcinoma, lung cancer, and osteoma [1]. The potential medicinal importance and our interest in the bioactive constituents prompted us to investigate the constituents of this plant. Previous studies indicated that phenolic acid compounds [2, 3], oleanane and ursane triterpenoids [4], are the main constituents contained in this plant. In this article, we describe the isolation and characterization of two oleanane triterpenoids 1, 2 and an octanordammarane triterpenoid 3.
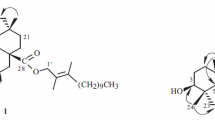
Compound 1 was isolated as a colorless needle-like crystal in CH3COCH3. The UV spectrum showed an absorption maximum at 248 nm, indicating the presence of a conjugated system. IR absorption bands indicated the existence of hydroxy (3524 and 3366 cm−1), γ-lactonic carbonyl (1774 cm−1), and olefinic (1667 cm−1) functional groups. It was assigned the molecular formula C30H44O4 with nine degrees of unsaturation, as deduced from the HR-ESI-MS (m/z 469.3332 [M + H]+, calcd 469.3318) and 13C NMR spectra. The 1H, 13C, and HSQC NMR data revealed the existence of seven methyl, methylene, methine, and nine quaternary carbons, including two cyclic olefinic bonds (δC 123.4, 129.5, 132.8, and 134.9) and one γ-lactonic carbonyl carbon (δC 178.4). The overall appearance of the 13C NMR spectrum showed the same planar structure as that of 2α,3β-dihydroxyolean-11,13(18)-dien-19β,28-olide [5] with an oleanane-type skeleton. The major difference was a 3α-hydroxy instead of the 3β-hydroxy. This was confirmed by NOESY experiments (Fig. 1). Strong NOE correlations between H-2 and H-24 and between H-3 and H-24 indicated that these protons were cofacial, and they were assigned a β-orientation.

Key HMBC and NOESY correlations of 1.
The observed oxygen-bearing tertiary carbon signal at C-19 (δ 85.3) as well as the HMBC correlations from H-19 (δ 4.72, s) to C-13, C-17, C-18, C-20, C-21, C-28, C-29 indicated a five-membered lactone ring between C-17 and C-19 which are also supported by IR absorption and nine degrees of unsaturation. The locations of the two double bonds at ∆11 and ∆13(18) were determined by the HMBC correlations from H-11 to C-8, C-9, C-10, and C-13, from H-12 to C-9, C14, C-13, and C-18, from H-27 to C-13, and from H-19 to C-13 and C-18. Therefore, compound 1 was designated as 2α,3α-dihydroxyolean-11,13(18)-dien-19β,28-olide.
Compound 2 was obtained as a white crystal in petroleum ether–ethyl acetate. Its molecular formula, C30H48O4, was determined from the HR-ESI-MS (m/z 495.3480 [M + Na]+, calcd 495.3450) analysis in the positive mode. The IR spectrum of 2 revealed stretchings for hydroxyl groups at 3734 and 3399 cm−1, acid group at 1696 cm−1, and olefin group at 1600 cm−1. The 1H, 13C, and HSQC NMR data demonstrated the presence of seven methyl, ten methylene, four methine, and nine quaternary carbons, including two olefinic quaternary carbons (δ 128.9 and 137.9), and a carboxylic acid carbon (δ 179.8). Attachment of a double bond to C-13 and C-18 was established by HMBC correlations from H-27 (δ 1.03) to C-13 (δ 137.9) and from H-19 (1.98 and 2.45) to C-18 (128.9). The NOESY correlations from H-2 to H-25 and from H-3 to H-24 indicated the β-orientation for H-2 and H-3. Thus, the structure of 2 was determined to be 2α,3α-dihydroxyolean-13(18)-en-28-oic acid. This is the first report of 2 from a natural source together with its full spectral data, although it has been prepared synthetically [6].
Compound 3 was obtained as white crystals with mp 210–211°C. It showed the [M]+ ion peak at m/z 334 in the EI-MS. The IR spectrum displayed hydroxyl groups at 3380 and 3262 cm−1. The 1H, 13C, and HSQC NMR data demonstrated the presence of five methyl, eight methylene, five methine (two of them oxygenated), and four quaternary carbons. Comparison of the 13C NMR data with those of octanordammarane triterpenoids [7] showed that it had a similar tetracyclic triterpenoid skeleton of dammarane type except for the absence of the signal for the side chain. The attachment of two hydroxyl groups to C-3 and C-17 was established by the HMBC correlations from H-3 (δ 3.19) to C-4 (39.1), C-23 (28.1), and C-24 (15.5) and from H-17 (3.90) to C-12 (24.3), C-13 (50.3), and C-15 (30.2). The NOESY correlations from H-3 to H-5 and from H-17 to H-30 indicated the α-orientation for H-3 and H-17 (Fig. 2). Therefore, compound 3 was designated as 3β-hydroxy- 20,21,22,23,24,25,26,27-octanordammaran-17β-ol.

Key HMBC and NOESY correlations of 3.
Considering that the roots have been used in folk medicine for the treatment of nasopharyngeal carcinoma, hepatic carcinoma, lung cancer, and osteoma, we evaluated the antitumor activities of compounds 2 and 3 (compound 1 was difficult to be dissolved in DMSO) against CNE-1 nasopharyngeal carcinoma, Spc gastric cancer, and HGC-27 lung cancer using the MTT assay; however, neither of them showed significant inhibitory activity (IC50 > 40 μg/mL for the three cell lines). No significant cytotoxicity was observed against Chang Liver normal cell line (IC50 > 100 μg/mL for the four cell lines).
Experimental
General Experimental Procedures. All melting points were recorded with a Shanghai SGW X-4 stage apparatus. UV spectrum was measured on a Hitachi U-3900 spectrophotometer. IR spectra were acquired using Spectrum 100 (PerkinElmer). NMR spectra were recorded on a Bruker AV III 500 MHz. HR-ESI-MS spectra were recorded on an ACQUITY UPLC/Q-TOF micro system (Waters Co., MA, USA). EI spectra were recorded on a DSQ mass spectrometer (Thermo).
Column chromatography was performed with silica gel (Marine Chemical Industry Factory, Qingdao, China) and Sephadex LH-20 (Merck, Darmstadt, Germany). TLC analyses were carried out on silica gel G plates with detection accomplished by spraying with 10% H2SO4 followed by heating at 105°C.
Plant Material. The roots and stems of Rubus alceaefolius Poir. were purchased from Guangzhou Kangsheng Corporation of Materia Medica, Guangdong Province, P. R. China, in February 2013, and identified by Ass. Prof. Zhijian Fang, Guangdong Pharmaceutical University.
Extraction and Isolation. The dried powdered stems and roots of Rubus alceaefolius Poir. (20 kg) were extracted with 95% ethanol under reflux (3 h, twice, at 80°C). The EtOH was evaporated under reduced pressure to yield a residue (1712 g). The residue was dissolved in 3000 mL water. The suspending solution was partitioned successively with PE, CHCl3, EtOAc, and n-butanol. After evaporation of the solvent, the PE partition solution yielded a residue (49.5 g), which was separated by column chromatography (1.2 kg silica gel, 100–200 mesh, gradient system PE–EtOAc) to yield fractions 1–10. Compound 1 (11 mg) was obtained by eluting Fr. 5 with PE–EtOAc (8:1). Fraction 3 was repeatedly separated on silica gel and purified by Sephadex LH-20 to afford compound 3 (10 mg). Fraction 8 was separated using PE–EtOAc (3:1) to afford compound 2 (42 mg).
Compound 1. C30H44O4. Colorless needle-like crystals (in CH3COCH3), mp 311–312°C. UV (MeOH, λmax, nm) (log ε): 248 (4.23). IR (KBr, ν, cm−1): 3524, 3366, 2927, 1774, 1667, 1453, 1386, 1233, 1026. For 1H and 13C NMR (CDCl3), see Tables 1 and 2. HR-ESI-MS m/z 469.3332 [M + H]+ (calcd for C30H45O4, 469.3318).
Compound 2. C30H48O4. White crystals (petroleum ether–ethyl acetate), mp 272–273°C. IR (KBr, ν, cm−1): 3734, 3399, 2963, 1696,1600, 1456, 1383, 1228, 1035, 762. For 1H and 13C NMR (Py-d5), see Tables 1 and 2. HR-ESI-MS m/z 495.3480 [M + Na]+ (calcd for C30H48O4Na, 495.3450).
Compound 3. C22H38O2. White crystals (in CH3COCH3), mp 210–211°C. IR (KBr, ν, cm−1): 3380, 3262, 2948, 1447, 1384, 1044, 750. For 1H and 13C NMR (CDCl3), see Tables 1 and 2. EI-MS m/z 334, 316, 207, 189, 97.
References
Q. X. Mei, The Study and countermeasure on the Nasopharyngeal Carcinoma [in Chinese], China Press of Traditional Chinese Medicine, Beijing, 2010, 28 pp.
C. B. Cui, Q. C. Zhao, B. Cai, X. S. Yao, and H. Osadsa, Asian Nat. Prod. Res., 4, 243 (2002).
C. B. Cui, Q. C. Zhao, B. Cai, X. S. Yao, and H. Osadsa, Chin. Chem. Lett., 4, 327 (2002).
L. Gan, Y. Zhao, J. Zhang, and F. Jiang, Chin. Mater. Med., 6, 362 (1998).
C. H. Lin, H. S. Chang, H. R. Liao, I. C. Chen, and I. L. Tsai, Int. J. Mol. Sci., 14, 8890 (2013).
H. B. Sun, P. Zhang, and J. Hao, Pat. CN 102050861 A, P.R. China, 2011.
J. Kitajima, K. Kimizuka, and Y. Tanaka, Chem. Pharm. Bull., 47, 1138 (1999).
Acknowledgment
This article is supported by the Science and Technology Foundation of Traditional Chinese Medicine Bureau of Guangdong Province (No. 20111083) and Zhongshan Science and Technology Foundation (No. 20113A007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 2, March–April, 2016, pp. 218–221.
Rights and permissions
About this article

Cite this article
Chen, P., Yan, H., Mei, Q. et al. Triterpenoids from the Roots and Stems of Rubus alceaefolius . Chem Nat Compd 52, 248–251 (2016). https://doi.org/10.1007/s10600-016-1606-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-016-1606-7