

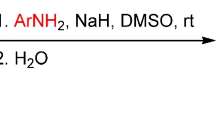
Direct SNH amidation of 5-, 6-, 7-, and 8-nitroquinolines in anhydrous DMSO was used to obtain the respective aroylamino derivatives of nitro- and nitrosoquinolines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The quinoline system is widely regarded as a privileged structure in the sense that many compounds containing quinoline ring as the key structural motif have shown a broad spectrum of biological and pharmaceutical activity and comprise a large category of natural alkaloids.1 There is still a considerable interest in searching for new synthetic routes that would provide access to derivatives of this heterocyclic system.2 The current possibilities of organic synthesis include direct C–H-functionalization of aromatic compounds, such as formation of C–N bonds,3 which can be performed in accordance with the principles of green chemistry, while achieving good atom economy.4
Direct C–H-functionalization of aromatic compounds can be currently performed by catalyzed or noncatalyzed reactions. Significant progress has been achieved with the first of these two options, where C–H bonds are activated in the presence of transition metal complexes as catalysts. This allows to selectively introduce amine and amide substituents in the molecules of electron-donating heterocycles. 5 However, this approach is problematic for the synthesis of active pharmaceutical ingredients6a and organic dyes for solar cells,6b since the presence of transition elements even in trace amounts is unacceptable for these applications.
Noncatalyzed C–H-functionalization can include, in particular, nucleophilic substitution of hydrogen (SNH) in the case of π-electron-poor substrates, such as π-electron-poor hetarenes and nitroarenes. Such reactions proceed 0mostly along two different routes: as vicarious or oxidative substitution.7 Both routes start from a nucleophilic addition step with the formation of a σH-adduct, followed by its aromatization either by the elimination of simple HX molecules, when a good leaving group X is bonded to the nucleophilic center, or by the action of external oxidant. The mechanism of dehydroaromatization in the presence of oxidant is affected both by the structure of the σН-complex, as well as the type of oxidant used, as well as the reaction conditions.7a,8 The most likely sequence of mechanistic steps leading to the aromatization includes a transfer of one electron, a proton, and another electron (the EPE mechanism) to the oxidant.8a In any case, the hydrogen atom is lost in the form of a proton, not a hydride ion.
The procedure for oxidative nucleophilic substitution of hydrogen does not require preliminary introduction of auxiliary or nucleofugic groups into the molecule of the substrate or reactant and does not require the use of costly catalysts or ligands. The oxidation of σH-adducts can be achieved by using organic or inorganic compounds, air oxygen,9,10 while stable intermediates can be oxidized electrochemically on anode.11 In the absence of an external oxidant, the NO2 group12 or C=N bond of substrate13 also can act as acceptors of hydride ion. The SNH methodology is already used on industrial scale14 and in many cases offer an attractive alternative to cross-coupling reactions in the presence of transition metals.15
The goal of this work was to study the possibilities for direct nucleophilic substitution of hydrogen with N-amide functionality in the molecules of nitroquinolines containing a nitro group in the benzene ring of the molecule. It is known that such compounds readily participate both in oxidative amination16 and arylamination reactions,17 as well as in vicarious SNH amination,18 while the regioselectivity of the process is determined solely by the presence of a nitro group.
In contrast to SNH amination processes, amidation reactions by direct substitution of hydrogen are still quite rare. Oxidative SNH amidation was first accomplished with nitrobenzene in 1993,19a even though an intramolecular variant of a similar reaction had been reported earlier.19b In a later study, the reaction of benzamide with 1,3-dinitrobenzene under anaerobic conditions gave a low yield of N-(2,4-dinitrophenyl)benzamide.12 Subsequent efforts at our laboratory led to successful oxidative SNH amidation of 1,3,7-triazapyrene,20 acridine,21 and 3-nitropyridine.22 The reactions in all cases were performed in anhydrous DMSO by treating the starting materials with separately prepared anions of the respective carboxamides at room temperature, using air oxygen20 or K3Fe(CN)621,22 in the role of oxidant.
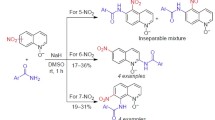
When studying the reaction of 5-nitroquinoline (1) with benzamide, we found that the optimal stoichiometry was 2 equiv of benzamide anion for 1 equiv of the starting material. The anions were generated separately by adding NaH to a solution of the respective benzamide in anhydrous DMSO at room temperature. After the addition of 5-nitroquinoline (1), the reaction reached completion in 2 h and gave a mixture of two products, which were separated by chromatography on silica gel. The products were identified as N-(5-nitroquinolin-8-yl)benzamide (2a) and N-(5-nitrosoquinolin-6-yl)benzamide (3a), with the overall yield of 66% (Scheme 1, Table 1, entry 1). The reaction proceeded in the presence of air oxygen, but the yield of nitro product 2a was only slightly lower when the reaction was performed under argon atmosphere (entry 2). Interestingly, the use of an external single-electron oxidant (K3Fe(CN)6) also was not very effective (entry 3). The obtained data indicate that, similarly to the case of 3-nitropyridine, 23 5-nitroquinoline exhibited dual reactivity, acting not only as a substrate, but also as the primary oxidant of σH-adducts during the formation of nitroamides 2. Such dual reactivity clearly reduced the yields of the target products and resulted in the presence of resinification products. We should also note that increasing the reaction temperature to 65–70°C accelerated the process (0.5 h), but improvement in the yield of nitroamide 2a to 22% was accompanied by sharp decrease in the yield of nitrosoamide 3a (entry 4).

Scheme 1
The anions of p-methyl- and p-methoxybenzamides reacted analogously, leading to the formation of the respective nitroamides 2b,с and nitrosoamides 3b,с with a significant predominance of the latter (Scheme 1, Table 1, entries 5, 6). However, p-nitrobenzamide gave not only the expected nitroamide 2d, but also its isomer at position 6 – 4-nitro-N-(5-nitroquinolin-6-yl)benzamide (4) (Scheme 1, Table 1, entry 7). It is likely that p-nitrobenzamide itself showed oxidizing properties toward the respective intermediates (o-nitrobenzamide formed a complex product mixture). Using amides of aliphatic acids (acetic, propionic, and isobutyric acids) was found to be ineffective, as it led to intractable product mixtures.
The formation of nitroso compounds 3a–c represents the first example of an alternative route for the aromatization of σH-adducts during oxidative SNH amidation reactions. This route competes with oxidative SNH reactions and is well known in the series of nitroarenes. It proceeds via dehydration of σH-intermediates to the respective nitroso compounds or products of their subsequent transformations. 2,7e,24 Similar reactions in the series of nitrohetarenes were recently observed during arylamination of 5-nitroindole24e and 3-nitropyridine,23a as well as upon carbamoylamination of the latter.23b In the case of amidation of 5-nitroquinoline (1), the nucleophile was added in the first step at the ortho and para positions relative to the NO2 group, while para-σH-adduct 5 underwent further oxidative aromatization with the formation of nitroamides 2a–d (Scheme 2, path a), but its ortho-analog 6 was aromatized by a sequence of steps including proton transfer and elimination of a water molecule, giving nitrosoamides 3a–c (Scheme 2, path b).

Scheme 2
A characteristic feature in 1H NMR spectra of nitrosoamides 3a–c in CDCl3 was the strong downfield shift of NH proton signals (13.5–13.6 ppm), providing evidence of a strong intramolecular NH···O=N hydrogen bond. In the case of their nitro analog 4, the intramolecular hydrogen bond was weaker (the NH proton signal was observed at δ 10.41 ppm). The structures of N-(5-nitrosoquinolin-6-yl)benzamide (3a) and 4-nitro-N-(5-nitroquinolin-6-yl)benzamide (4) were confirmed by X-ray structural analysis (Figs. 1, 2). According to X-ray diffraction data, the length of the O···HN hydrogen bond for nitro product 4 in crystalline state (2.098 Å) was substantially longer than for nitroso compound 3a (1.808 Å).

The molecular structure of compound 3a with atoms represented by thermal vibration ellipsoids of 50% probability. The intramolecular O···HN hydrogen bond (1.808 Å) is shown by a dotted line.

The molecular structure of compound 4 with atoms represented by thermal vibration ellipsoids of 50% probability. The intramolecular O···HN hydrogen bond (2.098 Å) is shown by a dotted line.
It is known that in the absence of a free para position relative to the NO2 group, as, for example, in the case of p-substituted nitrobenzenes24a or 5-nitroindole,24e the SNH arylamination leads only to the respective o-nitrosoamines or products of their subsequent transformations. Therefore, similar disproportionation products were also expected from the amidation of 6- and 7-nitroquinolines. Indeed, when using benzamide and its p-methyl and p-methoxy derivatives, the reaction with these nitroquinolines led exclusively to the formation of nitroso compounds in low or moderate yields, and only one of the two ortho positions relative to the nitro group was involved in the reaction. Thus, 6-nitroquinoline (7) participated in SNH amidation reaction exclusively at position 5, forming the respective N-(6-nitrosoquinolin-5-yl)benzamides 8a–с (Scheme 3, Table 2, entries 1–3), while 7-nitroquinoline (10) reacted at position 8, with the formation of N-(7-nitrosoquinolin-8-yl)- benzamides 11a–с (Scheme 3, Table 2, entries 5–7).

Scheme 3
However, similarly to the example of 5-nitroquinoline, the use of p-nitrobenzamide in both cases led to the respective dinitro compounds as amidation products: 4-nitro-N-(6-nitroquinolin-5-yl)benzamide (9) and 4-nitro-N-(7-nitroquinolin-8-yl)benzamide (12) (Scheme 3, Table 2, entries 4 and 8). It should be noted that compound 12 has been synthesized earlier by the nitration of N-(quinolin-8-yl)benzamide.25
Interestingly, in contrast to nitrosoamides 3a–c, the signals of NH protons in 1H NMR spectra of their analogs 8a–c and 11a–c in CDCl3 showed much weaker downfield shifts. In our opinion, the reason for this was steric hindrance by the hydrogen atom or lone electron pair of nitrogen atom at the peri positions, resulting in acoplanarity of the amide group and quinoline ring. Such interpretation was confirmed by X-ray structural analysis of 4-methoxy-N-(6-nitrosoquinolin-5-yl)benzamide (8c) (Fig. 3).

The molecular structure of compound 8c with atoms represented by thermal vibration ellipsoids of 50% probability.
We later found that, in contrast to the 5-, 6-, and 7-isomers (compounds 1, 7, and 10, respectively), SNH amidation of 8-nitroquinoline (13) under the same conditions proceeded exceedingly slowly and remained incomplete after 48 h. The use of K3Fe(CN)6 as external single-electron oxidant in this case was successful, even though a 6-fold excess of the amidating agent was necessary to complete the reaction, as well as the reaction duration was substantially longer (Table 2, entries 9–12). 8-Nitroquinoline (13) reacted with aromatic amide anions at the para position relative to the nitro group, forming products from oxidative nucleophilic substitution of hydrogen – N-(8-nitroquinolin-5-yl)benzamides 14a–d as the only reaction products (Table 2, entries 9–12).
The benzamide anions used in this study clearly were weaker nucleophiles than the NH2– anion, but were more nucleophilic than ammonia. Thus, the regioselectivity observed in oxidative SNH amination of 5-, 6-, and 7-nitroquinolines (compounds 1, 7, 10) in liquid NH3 –KMnO4 system16b was practically the same as the regioselectivity of SNH amidation. However, 8-nitroquinoline (13) could not be aminated under those conditions, while products 14a–d from its amidation were obtained by us in high yields.
We further found that our synthesized N-(5-nitrosoquinolin-6-yl)benzamides 3a–c were readily deacylated as a result of alcoholysis in MeOH–K2CO3 system at room temperature, forming the expected single product –5-nitrosoquinolin-6-amine (15) (Scheme 4).

Scheme 4
The presence of an NH2 group was observed from 1H NMR spectrum of compound 15 in CDCl3 as two one-proton signals at 11.94 and 5.54 ppm, the first of which belonged to the proton involved in strong intramolecular hydrogen bond. In our opinion, compound 15, similarly to amides 3a–c, is a convenient object for further functionalization of quinolines. However, it should be noted that analogs of compounds 3a–c – nitroso compounds 8a and 11a – reacted quite slowly under the same conditions, giving intractable product mixtures.
Taking into account the reversibility of the first step involving the addition of nucleophile, such contrasting results in the SNH amidation reaction of isomeric nitroquinolines can be explained by the different thermodynamic stability of the σH-adducts, as well as the ratio of their aromatization rates according to the two routes (the kinetic factor). In comparison to nitroarenes, the π-electron-poor pyridine ring certainly facilitated the addition step, but was not expected to substantially stabilize the σH-intermediate, since the delocalization of negative charge that is possible in the case of 6- and 8-nitroquinolines with the participation of the pyridine nitrogen atom would imply simultaneous disruption of aromaticity in two rings. As a polar aprotic solvent, DMSO does not substantially solvate anionic species, thus enhancing the nucleophilicity of arylamide ions, but not stabilizing the anionic intermediates. It is possible that the strongly polar nature of DMSO promoted the formation of more polar anionic σH-adducts, thus influencing the regioselectivity of the reaction.
Thus, the use of amide anions of aromatic acids as nucleophilic agents in the reaction with 8-nitroquinoline in anhydrous DMSO led to the formation of products from SNH amidation at position 5. Furthermore, 5-nitroquinoline formed a mixture of 6- and 8-aroylamino derivatives of 5-nitro- and 5-nitrosoquinoline. In the case of 6- and 7-isomers, the products were amides derived from 6- and 7-nitrosoquinolines, respectively. The regioselectivity of reactions in all cases was determined exclusively by the nitro group.
Experimental
IR spectra were recorded on a Shimadzu IRTracer-100 FTIR spectrometer for samples as thin films. 1H and 13C NMR spectra were acquired on a Bruker Avance HD 400 spectrometer (400 and 100 MHz, respectively). The internal standards were residual DMSO-d6 signals (2.50 ppm for 1H nuclei, 40.5 ppm for 13С nuclei)26 and TMS when CDCl3 was used as solvent. The structures of the key products (compounds 2a, 3a, 4, 12a) were confirmed by 2D NMR experiments (1H–1H COSY, 1H–13C HSQC, and 1H– 13C HMBC) on the same instrument (see the Supplementary information file). Mass spectra were recorded on a Bruker UHR-TOF maXis spectrometer (electrospray ionization). Melting points were determined on a REACH Devices RD-MP digital melting point apparatus. The reaction progress was controlled by TLC on Silufol UV-254 plates. The nitroquinolines and NaH (60% suspension in paraffin oil, abcr GmbH) were obtained from commercial sources and used without additional purification.
Amidation of 5-nitroquinoline (1). A solution of the appropriate amide (1 mmol) in anhydrous DMSO (4 ml) was treated at room temperature by adding a suspension of NaH in paraffin oil (40 mg, 1 mmol of NaH) and 5-nitroquinoline (1) (87 mg, 0.5 mmol). The mixture was vigorously stirred at room temperature for the duration indicated in Table 1. The reaction mixture was then poured onto ice (50 g) and after warming to room temperature was acidified with dilute HCl solution to pH ~7. The precipitate that formed was filtered off, washed with water, and dried. The obtained mixture was separated into fractions by the dry silica gel flash chromatography.27
N-(5-Nitroquinolin-8-yl)benzamide (2a). The first yellow fraction, eluent PhH. Yield 18 mg (12%), pale- yellow crystals, mp 213–214°C (decomp., PhH – petroleum ether) (mp 215–216°C28a, 209–210°C28b, 212–213°C28c). IR spectrum, ν, cm–1: 3356, 2921, 1687, 1498, 1382. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.50–7.67 (3H, m, H-3,4,5 Ph); 7.77 (1H, dd, J = 8.8, J = 4.1, H-3); 8.10 (2H, d, J = 7.6, H-2,6 Ph); 8.63 (1H, d, J = 8.8, H-6); 8.97 (1H, d, J = 4.1, H-2); 9.03 (1H, d, J = 8.8, H-7); 9.32 (1H, d, J = 8.8, H-4); 11.10 (1H, br. s, NH). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.66 (2H, br. d, J = 7.3, H-3,5 Ph) 7.72 (1H, br. t, J = 7.1, H-4 Ph); 7.97 (1H, dd, J = 8.6, J = 4.4, H-3); 8.07 (2H, br. d, J = 7.7, H-2,6 Ph); 8.67 (1H, d, J = 8.8, H-6); 8.87 (1H, d, J = 8.8, H-7); 9.11–9.17 (2H, m, H-2,4); 11.03 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 113.9 (C-7); 122.0 (C-4a); 124.9 (C-3); 127.6 (C-2,6 Ph); 128.1 (C-6); 129.2 (C-3,5 Ph); 132.8 (C-4 Ph); 133.6 (C-4); 134.3 (C-1 Ph); 137.9 (C-8a); 138.8 (C-5); 141.0 (C-8); 149.2 (C-2); 165.9 (C=O). Found, m/z: 316.0701 [М+Na]+. C16H11N3NaO3. Calculated, m/z: 316.0693.
N-(5-Nitrosoquinolin-6-yl)benzamide (3a). The second yellow fraction, eluent PhH–EtOAc, 5:1. Yield 75 mg (54%), green crystals, mp 181–182°C (decomp., PhH – petroleum ether). IR spectrum, ν, cm–1: 3059, 1694, 1585, 1498, 1354. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.63–7.73 (3H, m, H-3,4,5 Ph); 7.81 (1H, dd, J = 8.6, J = 4.2, H-3); 8.20 (2H, d, J = 7.7, H-2,6 Ph); 8.53 (1H, d, J = 9.6, H-8); 9.05 (1H, d, J = 4.2, H-2); 9.39 (1H, d, J = 9.6, H-7); 9.80 (1H, d, J = 8.6, H-4); 13.45 (1H, br. s, NH). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.67– 7.80 (3H, m, H-3,4,5 Ph); 7.94 (1H, dd, J = 8.5, J = 4.1, H-3); 8.14 (2H, br. d, J = 6.8, H-2,6 Ph); 8.60 (1H, d, J = 9.4, H-8); 9.03–9.10 (2H, m, H-2,7); 9.49 (1H, br. d, J = 8.2, H-4); 12.81 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 122.9 (C-7); 124.6 (C-4a); 126.5 (C-6); 126.7 (C-3); 128.0 (C-2,6 Ph); 129.3 (C-3,5 Ph); 130.7 (C-4); 133.3 (C-1,4 Ph); 143.1 (C-8); 143.6 (C-8a); 149.0 (C-5); 151.0 (C-2); 167.6 (C=O). Found, m/z: 300.0749 [М+Na]+. C16H11N3NaO2. Calculated, m/z: 300.0743.
4-Methyl-N-(5-nitroquinolin-8-yl)benzamide (2b). The first pale-yellow fraction, eluent PhH. Yield 20 mg (13%), pale-yellow crystals, mp 245–246°C (decomp., PhH – petroleum ether) (mp 210–211°C28b). IR spectrum, ν, cm–1: 3348, 1689, 1570, 1504, 1394. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 2.47 (3H, s, CH3); 7.38 (2H, d, J = 8.1, H-3,5 Ar); 7.76 (1H, dd, J = 8.8, J = 4.2, H-3); 7.99 (2H, d, J = 8.1, H-2,6 Ar); 8.61 (1H, d, J = 8.8, H-7); 8.96 (1H, dd, J = 4.2, J = 1.4, H-2); 9.01 (1H, d, J = 8.8, H-6); 9.31 (1H, dd, J = 8.8, J = 1.4, H-4); 11.06 (1H, br. s, NH). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 2.44 (3H, s, CH3); 7.46 (2H, d, J = 7.8, H-3,5 Ar); 7.95–8.00 (3H, m, H-2,6 Ar, H-3); 8.66 (1H, d, J = 8.8, H-7); 8.87 (1H, d, J = 8.8, H-6); 9.11–9.17 (2H, m, H-2,4); 10.98 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 20.9; 113.3; 120.9; 125.2; 127.1; 127.3; 129.5; 130.8; 132.7; 137.0; 138.4; 140.2; 142.9; 149.9; 164.7. Found, m/z: 330.0856 [М+Na]+. C17H13N3NaO3. Calculated, m/z: 330.0849.
4-Methyl-N-(5-nitrosoquinolin-6-yl)benzamide (3b). The second yellow fraction, eluent PhH–EtOAc, 5:1. Yield 103 mg (71%), yellowish-green crystals, mp 170–171°C (decomp., PhH – petroleum ether). IR spectrum, ν, cm–1: 3360, 3036, 1688, 1580, 1347. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 2.50 (3H, s, CH3); 7.45 (2H, d, J = 8.0, H-3,5 Ar); 7.80 (1H, dd, J = 8.5, J = 4.2, H-3); 8.10 (2H, d, J = 8.0, H-2,6 Ar); 8.52 (1H, J = 9.6, H-8); 9.03 (1H, d, J = 4.2, H-2); 9.39 (1H, d, J = 9.6, H-7); 9.79 (1H, d, J = 8.5, H-4); 13.49 (1H, br. s, NH). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 2.45 (3H, s, CH3); 7.51 (2H, d, J = 8.0, H-3,5 Ar); 7.93 (1H, dd, J = 8.5, J = 4.1, H-3); 8.03 (2H, d, J = 8.0, H-2,6 Ar); 8.58 (1H, d, J = 9.5, H-8); 9.03 (1H, d, J = 4.1, H-2); 9.08 (1H, d, J = 9.5, H-7); 9.50 (1H, d, J = 8.5, H-4); 12.87 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 21.2; 122.7; 125.2; 126.0; 126.6; 128.1; 129.9; 130.4; 130.7; 143.2; 143.5; 143.8; 148.9; 151.0; 167.5. Found, m/z: 314.0901 [М+Na]+. C17H13N3NaO2. Calculated, m/z: 314.0900.
4-Methoxy-N-(5-nitroquinolin-8-yl)benzamide (2c).25,28d,e The first pale-yellow fraction, eluent PhH. Yield 31 mg (19%), pale-yellow crystals, mp 255–256°C (decomp., PhH – petroleum ether). IR spectrum, ν, cm–1: 3326, 2854, 1680, 1502, 1350. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 3.85 (3H, s, OCH3); 7.01 (2H, d, J = 8.5, H-3,5 Ar); 7.70 (1H, dd, J = 8.7, J = 4.0, H-3); 8.01 (2H, d, J = 8.5, H-2,6 Ar); 8.56 (1H, d, J = 8.8, H-7); 8.90 (1H, d, J = 4.0, H-2); 8.94 (1H, d, J = 8.8, H-8); 9.26 (1H, d, J = 8.7, H-4); 10.97 (1H, br. s, NH). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 3.88 (3H, s, OCH3); 7.19 (2H, d, J = 8.5, H-3,5 Ar); 7.99 (1H, dd, J = 8.5, J = 4.4, H-3); 8.06 (2H, d, J = 8.5, H-2,6 Ar); 8.68 (1H, d, J = 8.9, H-7); 8.87 (1H, d, J = 8.9, H-8); 9.12–9.18 (2H, m, H-2,4); 10.98 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 54.5; 112.5; 113.2; 120.9; 123.7; 125.3; 127.1; 128.5; 132.4; 136.7; 137.4; 140.1; 148.0; 162.1; 164.2. Found, m/z: 346.0804 [М+Na]+. C17H13N3NaO4. Calculated, m/z: 346.0798.
4-Methoxy-N-(5-nitrosoquinolin-8-yl)benzamide (3c). The second yellow fraction, eluent PhH–EtOAc, 5:1. Yield 80 mg (52%), orange crystals, mp 185–186°C (decomp., PhH – petroleum ether). IR spectrum, ν, cm–1: 3366, 3069, 1688, 1585, 1357. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 3.95 (3H, s, OCH3); 7.12 (2H, d, J = 8.6, H-2,6 Ar); 7.78 (1H, dd, J = 8.5, J = 4.1, H-3); 8.18 (2H, d, J = 8.6, H-3,5 Ar); 8.49 (1H, d, J = 9.6, H-8); 9.03 (1H, d, J = 4.1, H-2); 9.37 (1H, d, J = 9.6, H-7); 9.78 (1H, d, J = 8.5, H-4); 13.56 (1H, br. s, NH). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 3.91 (3H, s, OCH3); 7.25 (2H, d, J = 8.8, H-2,6 Ar); 7.94 (1H, dd, J = 8.5, J = 4.1, H-3); 8.12 (2H, d, J = 8.8, H-3,5 Ar); 8.58 (1H, d, J = 9.5, H-8); 9.03 (1H, dd, J = 4.1, J = 1.2, H-2); 9.09 (1H, d, J = 9.5, H-7); 9.53 (1H, br. d, J = 8.5, H-4); 12.97 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 56.2; 115.1; 123.1; 125.6; 126.0; 126.4; 126.9; 130.7; 131.0; 143.7; 143.9; 149.3; 151.3; 163.7; 167.4. Found, m/z: 308.1037 [М+H]+. C17H14N3O3. Calculated, m/z: 308.1030.
4-Nitro-N-(5-nitroquinolin-8-yl)benzamide (2d).25 The first light-yellow fraction, eluent PhH. Yield 12 mg (7%), light-brown crystals, mp 264–265°C (decomp., PhH – petroleum ether). IR spectrum, ν, cm–1: 3311, 3114, 1687, 1538, 1305. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.81 (1H, dd, J = 8.8, J = 4.2, H-3); 8.27 (2H, d, J = 8.8, H-2,6 Ar); 8.45 (2H, d, J = 8.8, H-3,5 Ar); 8.64 (1H, d, J = 8.8, H-7); 8.99 (1H, dd, J = 4.2, J = 1.5, H-2); 9.01 (1H, d, J = 8.8, H-6); 9.33 (1H, dd, J = 8.8, J = 1.5, H-4); 11.15 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 114.3; 121.9; 124.4; 125.1; 127.8; 128.8; 133.8; 137.9; 139.7; 140.1; 149.5; 150.3; 160.3; 163.7. Found, m/z: 339.0714 [М+H]+. C16H11N4O5. Calculated, m/z: 339.0724.
4-Nitro-N-(5-nitroquinolin-6-yl)benzamide (4). The second yellow fraction, eluent PhH–EtOAc, 5:1. Yield 78 mg (46%), yellow crystals, mp 234–235°C (decomp., PhH – petroleum ether). IR spectrum, ν, cm–1: 3368, 3105, 1692, 1588, 1418. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.63 (1H, dd, J = 8.8, J = 4.2, H-3); 8.17 (2H, d, J = 6.9, H-2,6 Ar); 8.40–8.44 (3H, m, H-3,5 Ar, H-8); 8.58 (1H, dd, J = 8.8, J = 1.5, H-4); 8.95 (1H, d, J = 9.4, H-7); 8.99 (1H, dd, J = 4.2, J = 1.5, H-2); 10.41 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 121.9 (C-4a); 123.7 (C-7); 124.3 (C-3); 124.5 (C-3,5 Ar); 128.9 (C-2,6 Ar); 131.4 (C-4); 132.5 (C-6); 134.5 (C-5); 136.7 (C-8); 138.9 (C-1 Ar); 144.9 (C-8a); 150.5 (C-4 Ar); 151.1 (C-2); 163.8 (CO). Found, m/z: 339.0727 [М+H]+. C16H11N4O5. Calculated, m/z: 339.0724. Found, m/z: 361.0533 [М+Na]+. C16H10N4NaO5. Calculated, m/z: 361.0543.
Amidation of 6- and 7-nitroquinolines 7, 10 (General method). A solution of the appropriate amide (0.75 mmol) in anhydrous DMSO (4 ml) was stirred at room temperature and treated by adding NaH suspension in paraffin oil (30 mg, 0.75 mmol NaH) and 6- or 7-nitroquinoline (87 mg, 0.5 mmol, compounds 7 or 10, respectively). The mixture was vigorously stirred at room temperature for the duration indicated in Table 2. The reaction mixture was then poured onto ground ice (50 g) and after warming to room temperature was acidified with dilute HCl solution to pH ~7. The precipitate that formed was filtered off, washed with water, and dried. The product was recrystallized from a suitable solvent.
N-(6-Nitrosoquinolin-5-yl)benzamide (8a). Yield 101 mg (73%), light-green crystals, mp 175–176°C (decomp., EtOAc – petroleum ether). IR spectrum, ν, cm–1: 3233, 1657, 1613, 1500, 1386. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 6.95 (1H, d, J = 9.2, H-8); 7.55–7.62 (3H, m, H-3, H-3,5 Ph); 7.68 (1H, t, J = 7.4, H-4 Ph); 7.88 (1H, d, J = 9.2, H-7); 8.15 (2H, d, J = 7.4, H-2,6 Ph); 8.71 (1H, br. d, J = 8.5, H-4); 9.11 (1H, dd, J = 4.2, J = 1.5, H-2); 10.74 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 110.4; 122.0; 124.6; 128.1; 128.6; 129.3; 133.3 (2C); 136.7; 142.8; 152.4; 154.9; 155.5; 167.3. Found, m/z: 300.0737 [М+Na]+. C16H11N3NaO2. Calculated, m/z: 300.0743.
4-Methyl-N-(6-nitrosoquinolin-5-yl)benzamide (8b). Yield 97 mg (67%), yellowish-green crystals, mp 167–168°C (decomp., PhH). IR spectrum, ν, cm–1: 3301, 1671, 1537, 1493, 1321. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 2.48 (3H, s, CH3); 6.95 (1H, d, J = 9.2, H-8); 7.38 (2H, d, J = 8.0, H-3,5 Ar); 7.58 (1H, dd, J = 8.5, J = 4.2, H-3); 7.87 (1H, d, J = 9.2, H-7); 8.04 (2H, d, J = 8.0, H-2,6 Ar); 8.70 (1H, br. d, J = 8.5, H-4); 9.10 (1H, br. d, J = 4.2, H-2); 10.75 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 21.8; 110.5; 121.9; 124.6; 128.1; 128.4; 129.9; 130.4; 136.9; 143.0; 144.2; 152.4; 154.8; 155.6; 167.2. Found, m/z: 314.0916 [М+Na]+. C17H13N3NaO2. Calculated, m/z: 314.0900.
4-Methoxy-N-(6-nitrosoquinolin-5-yl)benzamide (8c). Yield 118 mg (77%), light-green crystals, mp 188–189°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3083, 2929, 1632, 1578, 1381. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 3.92 (3H, s, OCH3); 6.97 (1H, d, J = 9.2, H-8); 7.06 (2H, d, J = 8.7, H-2,6 Ar); 7.57 (1H, dd, J = 8.6, J = 4.2, H-3); 7.86 (1H, d, J = 9.2, H-7); 8.12 (2H, d, J = 8.7, H-3,5 Ar); 8.70 (1H, br. d, J = 8.6, H-4); 9.11 (1H, br. d, J = 4.2, H-2); 10.75 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 55.8; 110.6; 114.5; 121.8; 124.6; 125.4; 128.2; 130.2; 137.0; 143.1; 152.4; 154.8; 155.6; 163.7; 166.8. Found, m/z: 330.0856 [М+Na]+. C17H13N3NaO3. Calculated, m/z: 330.0849.
4-Nitro-N-(6-nitroquinolin-5-yl)benzamide (9). The second yellow fraction, eluent PhH–EtOAc, 5:1. Yield 73 mg (43%), yellow crystals, mp 242–243°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3240, 1661, 1600, 1514, 1345. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.59 (1H, dd, J = 8.7, J = 4.2, H-3); 8.20 (1H, d, J = 9.4, H-8); 8.25 (2H, d, J = 8.7, H-2,6 Ar); 8.36 (1H, br. d, J = 8.7, H-4); 8.40 (1H, d, J = 9.4, H-7); 8.45 (2H, d, J = 8.7, H-3,5 Ar); 9.13 (1H, br. d, J = 4.2, H-2); 10.14 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 122.5; 123.9, 124.3; 124.5; 129.1; 129.9; 130.8; 135.7; 138.3; 140.3; 150.1; 150.6; 154.3; 164.7. Found, m/z: 361.0537 [М+Na]+. C16H10N4NaO5. Calculated, m/z: 361.0543.
N-(7-Nitrosoquinolin-8-yl)benzamide (11a). Yield 104 mg (75%), beige crystals, mp 159–160°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3317, 2921, 1684, 1510, 1408. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 6.77 (1H, d, J = 9.0, H-5); 7.51 (1H, d, J = 9.0, H-6); 7.56–7.61 (2H, m, H-3,5 Ph); 7.62–7.67 (2H, m, H-3, H-4 Ph); 8.20 (1H, br. d, J = 8.8, H-4); 8.22 (2H, d, J = 8.2, H-2,6 Ph); 9.00 (1H, br. d, J = 4.1, H-2); 10.42 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 109.1; 123.3; 124.8; 128.3; 129.0; 131.7; 132.9; 134.0; 136.7; 139.7; 142.7; 150.5; 152.5; 168.8. Found, m/z: 300.0746 [М+Na]+. C16H11N3NaO2. Calculated, m/z: 300.0743.
4-Methyl-N-(7-nitrosoquinolin-8-yl)benzamide (11b). Yield 108 mg (74%), yellowish-green crystals, mp 169– 170°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3297, 1682, 1516, 1480, 1397. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 2.48 (3H, s, CH3); 6.77 (1H, d, J = 9.0, H-5); 7.38 (2H, d, J = 8.2, H-3,5 Ar); 7.49 (1H, d, J = 9.0, H-6); 7.64 (1H, dd, J = 8.3, J = 4.2, H-3); 8.12 (2H, d, J = 8.2, H-2,6 Ar); 8.19 (1H, dd, J = 8.3, J = 1.6, H-4); 8.99 (1H, dd, J = 4.2, J = 1.6, H-2); 10.41 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 21.8; 109.2; 123.1; 124.7; 128.3; 129.7; 131.1; 131.7; 136.7; 139.8; 142.7; 143.6; 150.4; 152.5; 168.7. Found, m/z: 314.0896 [М+Na]+. C17H13N3NaO2. Calculated, m/z: 314.0900.
4-Methoxy-N-(7-nitrosoquinolin-8-yl)benzamide (11c). Yield 123 mg (80%), yellowish-green crystals, mp 165–166°C (decomp., PhH). IR spectrum, ν, cm–1: 3320, 3078, 1682, 1505, 1395. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 3.92 (3H, s, OCH3); 6.76 (1H, d, J = 9.0, H-5); 7.06 (2H, d, J = 8.7, H-2,6 Ar); 7.47 (1H, d, J = 9.0, H-6); 7.62 (1H, dd, J = 8.2, J = 4.2, H-3); 8.17–8.19 (3H, m, H-4, H-3,5 Ar); 8.98 (1H, br. d, J = 4.2, H-2); 10.37 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 55.7; 109.2; 114.2; 123.0; 124.7; 126.1; 130.3; 131.7; 136.7; 140.0; 142.7; 150.3; 152.5; 163.4; 168.3. Found, m/z: 330.0857 [М+Na]+. C17H13N3NaO3. Calculated, m/z: 330.0849.
4-Nitro-N-(7-nitroquinolin-8-yl)benzamide (12).25 The first yellow fraction, eluent 1:1 petroleum ether – EtOAc mixture. Yield 66 mg (39%), yellow crystals, mp 255–256°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3290, 3074, 1697, 1524, 1343. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.68 (1H, dd, J = 8.3, J =4.2, H-3); 7.76 (1H, d, J = 9.1, H-5); 8.08 (1H, d, J = 9.1, H-6); 8.27 (2H, d, J = 8.6, H-2,6 Ar); 8.31 (1H, br. d, J = 8.3, H-4); 8.41 (2H, d, J = 8.6, H-3,5 Ar); 8.98 (1H, br. d, J = 4.2, H-2); 10.45 (1H, br. s, NH). 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.76 (1H, d, J = 9.1, H-5); 7.83 (1H, dd, J = 8.3, J = 4.1, H-3); 8.14 (2H, s, H-5,6); 8.08 (1H, d, J = 9.1, H-6); 8.33 (2H, d, J = 8.8, H-2,6 Ar); 8.43 (2H, d, J = 8.8, H-3,5 Ar); 8.61 (1H, dd, J = 8.3, J = 1.5, H-4); 9.13 (1H, dd, J = 4.1, J = 1.5, H-2); 11.30 (1H, br. s, NH). 13C NMR spectrum (CDCl3), δ, ppm: 122.4; 123.9; 124.3; 124.4; 127.6; 129.3; 129.8; 136.8; 138.9; 140.5; 141.0; 150.4; 150.8; 163.5. Found, m/z: 361.0532 [М+Na]+. C16H10N4NaO5. Calculated, m/z: 361.0543.
Amidation of 8-nitroquinoline (13). A solution of the appropriate amide (3 mmol) in anhydrous DMSO (4 ml) was stirred at room temperature and treated with a suspension of NaH in paraffin oil (120 mg, 3 mmol NaH), 8-nitroquinoline (13) (87 mg, 0.5 mmol), and K3Fe(CN)6 (987 mg, 3 mmol). The mixture was vigorously stirred at room temperature for the duration indicated in Table 2. The crude products were further purified by recrystallization from EtOAc.
N-(8-Nitroquinolin-5-yl)benzamide (14a). Yield 92 mg (63%), beige crystals, mp 233–234°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3268, 3068, 1652, 1519, 1390. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.59– 7.61 (2H, m, H-3,5 Ph); 7.67 (1H, t, J = 7.2, H-4 Ph); 7.76 (1H, dd, J = 8.6, J = 4.1, H-3); 7.95 (1H, d, J = 8.2, H-6); 8.11 (2H, d, J = 7.7, H-2,6 Ph); 8.35 (1H, d, J = 8.2, H-7); 8.65 (1H, dd, J = 8.6, J = 1.5, H-4); 9.08 (1H, dd, J = 4.1, J = 1.5, H-2); 10.85 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 121.5 (C-6); 122.7 (C-3); 123.5 (C-7); 123.9 (C-4a); 128.1 (C-2,6 Ph); 128.6 (C-3,5 Ph); 132.2 (C-4 Ph); 133.1 (C-4); 133.9 (C-1' Ph); 137.8 (C-5); 139.1 (C-8a); 145.4 (C-8); 152.7 (C-2); 166.6 (C=O). Found, m/z: 316.0702 [М+Na]+. C16H11N3NaO3. Calculated, m/z: 316.0693.
4-Methyl-N-(8-nitroquinolin-5-yl)benzamide (14b). Yield 120 mg (78%), beige crystals, mp 234–235°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3263, 3050, 1649, 1527, 1360. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 2.42 (3H, s, CH3); 7.40 (2H, d, J = 8.0, H-3,5 Ar); 7.75 (1H, dd, J = 8.6, J = 4.0, H-3); 7.95 (1H, d, J = 8.2, H-6); 8.01 (2H, d, J = 8.0, H-2,6 Ar); 8.34 (1H, d, J = 8.2, H-7); 8.63 (1H, br. d, J = 8.6, H-4); 9.05 (1H, br. d, J = 4.0, H-2); 10.76 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 21.1; 121.5; 122.6; 123.6; 123.9; 128.2; 129.1; 131.0; 133.1; 138.0; 139.1; 142.3; 145.2; 152.7; 166.4. Found, m/z: 330.0858 [М+Na]+. C17H13N3NaO3. Calculated, m/z: 330.0849.
4-Methoxy-N-(8-nitroquinolin-5-yl)benzamide (14c). Yield 116 mg (72%), beige crystals, mp 235–236°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3262, 1649, 1604, 1530, 1381. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 3.87 (3H, s, OCH3); 7.12 (2H, d, J = 8.7, H-3,5 Ar); 7.75 (1H, dd, J = 8.6, J = 4.1, H-3); 7.92 (1H, d, J = 8.2, H-6); 8.10 (2H, d, J = 8.7, H-2,6 Ar); 8.33 (1H, d, J = 8.2, H-7); 8.62 (1H, br. d, J = 8.6, H-4); 9.06 (1H, br. d, J = 3.8, H-2); 10.68 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 55.6; 113.8; 121.4; 122.6; 123.6; 123.9; 125.9; 130.2; 133.2; 138.1; 139.1; 145.2; 152.7; 162.4; 165.9. Found, m/z: 346.0783 [М+Na]+. C17H13N3NaO4. Calculated, m/z: 346.0798.
4-Nitro-N-(8-nitroquinolin-5-yl)benzamide (14d). Yield 142 mg (84%), yellow crystals, mp 247–248°C (decomp., EtOAc). IR spectrum, ν, cm–1: 3424, 2924, 1693, 1516, 1345. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.77 (1H, dd, J = 8.7, J = 4.2, H-3); 7.98 (1H, d, J = 8.2, H-6); 8.33 (2H, d, J = 8.8, H-2,6 Ar); 8.36 (1H, d, J = 8.2, H-7); 8.43 (2H, d, J = 8.8, H-3,5 Ar); 8.69 (1H, br. d, J = 8.7, H-4); 9.09 (1H, br. d, J = 3.6, J = 1.3, H-2); 11.14 (1H, br. s, NH). 13C NMR spectrum (DMSO-d6), δ, ppm: 121.8; 122.8; 123.5; 123.7; 123.9; 129.7; 133.1; 137.2; 139.0; 139.6; 145.7; 149.5; 152.8; 165.1. Found, m/z: 361.0548 [М+Na]+. C16H10N4NaO5. Calculated, m/z: 361.0543.
Alcoholysis of nitroso compounds 3a–c (General method). A solution of the appropriate amide 3a–c (0.3 mmol) in MeOH (15 ml) was treated by adding K2CO3 (248.4 mg, 1.8 mmol), followed by vigorous stirring for 0.5 h. The mixture was then poured into cold water (100 ml) and acidified with dilute HCl solution to pH ~7. The product was extracted with EtOAc (3×15 ml), and the solvent was evaporated to dryness at reduced pressure. Further purification was performed by the dry silica gel flash chromatography,27 eluting with 5:1 PhH–EtOAc mixture and collecting the first (yellowish-green) fraction. After the removal of solvent, pure nitroso product 15 was obtained.
5-Nitrosoquinolin-6-amine (15). Yield 33 mg (64%, from compound 3a), 41 mg (79%, from compound 3b), 36 mg (71%, from compound 3c), green crystals, mp 190– 191°C (decomp., PhH) (mp 176°C29). IR spectrum, ν, cm–1: 3248, 2923, 1628, 1505, 1297. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 5.54 (1H, br. s, NH); 7.03 (1H, d, J = 9.4, H-8); 7.63 (1H, dd, J = 8.4, J = 4.3, H-3); 8.06 (1H, d, J = 9.4, H-7); 8.84 (1H, dd, J = 4.3, J = 1.5, H-2); 9.59 (1H, d, J = 8.4, H-4); 11.94 (1H, br. s, NH···O). 13C NMR spectrum (CDCl3), δ, ppm: 123.1; 124.7; 129.5; 130.7; 132.9; 142.2; 142.6; 148.6; 149.1. Found, m/z: 174.0654 [М+H]+. C9H8N3O. Calculated, m/z: 174.0662.
X-ray structural analysis of compounds 3a, 4, and 8с was performed on an Agilent SuperNova diffractometer equipped with a microfocus X-ray source containing a copper anode and Atlas S2 CCD matrix detector. Crystals suitable for X-ray structural analysis were obtained by slow evaporation of a solution in 1:1 petroleum ether – CH2Cl2 system (compound 3a), EtOAc (compound 4), or MeOH (compound 8с) at room temperature. The reflections were collected, unit cell parameters were determined and refined by using the specialized CrysAlisPro 1.171.38.41 software suite (Rigaku Oxford Diffraction, 2015).30 The structures were solved using ShelXT program (Sheldrick, 2015)31 and refined with ShelXL program (Sheldrick, 2015),32 Molecular graphics were rendered and prepared for publication using the Olex2 version 1.2.10 software suite.33 The complete X-ray diffraction datasets were deposited at the Cambridge Crystallographic Data Center (deposits CCDC 1871005 (compound 3a), CCDC 1874185 (compound 4), and CCDC 1904646 (compound 8с)).
Supplementary information file containing 1Н and 13С NMR spectra of all synthesized compounds is available from the journal website at http://springerlink.bibliotecabuap.elogim.com/journal/10593. This work received financial support from the Ministry of Education and Science of the Russian Federation within the framework of State Assignment (project No. 4.6306.2017/8.9).
References
(a) Michael, J. P. Nat. Prod. Rep. 1997, 14, 605. (b) Kumar, S.; Bawa, S.; Gupta, H. Mini-Rev. Med. Chem. 2009, 9, 1648. (c) Puskullu, M. O.; Tekiner, B.; Suzen, S. Mini-Rev. Med. Chem. 2013, 13, 365. (d) Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845. (e) Gopaul, K.; Shintre, S. A.; Koorbanally, N. A. Anticancer Agents Med. Chem. 2015, 15, 631. (f) Hussaini, S. M. Expert Opin. Ther. Pat. 2016, 26, 1201. (g) Jain, S.; Chandra, V.; Jain, P. K.; Pathak, K.; Pathak, D.; Vaidya, A. Arab. J. Chem. 2016. DOI:https://doi.org/10.1016/j.arabjc.2016.10.009. (h) Sharma, V.; Mehta, D. K.; Das, R. Mini-Rev. Med. Chem. 2017, 17, 1557. (i) Musiol, R. Expert Opin. Drug Discovery 2017, 12, 583.
Mąkosza, M.; Wojciechowski, K. Top. Heterocycl. Chem. 2014, 37, 51.
(a) Baeten, M.; Maes, B. U.W. Adv. Organomet. Chem. 2017, 67, 401. (b) C–H Bond Activation and Catalytic Functionalization. I and II; Dixneuf, P. H.; Doucet, H., Eds.; Springer: Berlin, 2016. (c) Metal Free C–H Functionalization of Aromatics. Nucleophilic Displacement of Hydrogen; Charushin, V.; Chupakhin, O., Eds.; Springer: Cham, 2014.
(a) Arends, I.; Sheldon, R.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, 2007. (b) Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411. (c) Utepova, I. A.; Trestsova, M. A.; Chupakhin, O. N.; Charushin, V. N.; Rempel, A. A. Green Chem. 2015, 17, 4401. (d) Lancaster, M. Green Chemistry. An Introductory Text; 2nd ed.; RSC Publishing: Cambridge, 2010. (e) Sheldon, R. A. Chem. Soc. Rev. 2012, 41, 1437.
(a) Kim, J.; Kim, J.; Chang, S. Chem.–Eur. J. 2013, 19, 7328. (b) Ryu, J.; Shin, K.; Park, S. H.; Kim, J. Y.; Chang, S. Angew. Chem. 2012, 124, 10042. (c) Shi, J.; Zhou, B.; Yang, Y.; Li, Y. Org. Biomol. Chem. 2012, 10, 8953.
(a) Verbitskiy, E. V.; Cheprakova, E. M.; Slepukhin, P. A.; Kravchenko, M. A.; Skornyakov, S. N.; Rusinov, G. L.; Chupakhin, O. N.; Charushin, V. N. Eur. J. Med. Chem. 2015, 97, 225. (b) Verbitskiy, E. V.; Cheprakova, E. M.; Subbotina, J. O.; Schepochkin, A. V.; Slepukhin, P. A.; Rusinov, G. L.; Charushin, V. N.; Chupakhin, O. N.; Makarova, N. I.; Metelitsa, A. V.; Minkin, V. I. Dyes Pigm. 2014, 100, 201.
(a) Chupakhin, O. N.; Charushin, V. N.; van der Plas, H. C. Nucleophilic Aromatic Substitution of Hydrogen; Academic Press: San Diego, 1994. (b) Chupakhin, O. N.; Charushin, V. N. Tetrahedron Lett. 2016, 57, 2665. (c) Charushin, V. N.; Chupakhin, O. N. Top. Heterocycl. Chem. 2014, 37, 1. (d) Gulevskaya, A. V.; Pozharskii, A. F. Top. Heterocycl. Chem. 2014, 37, 179. (e) Mąkosza, M. Synthesis 2017, 3247. (f) Suwiński, J. W. ARKIVOC 2017, (i), 402. (g) Czaban-Jóźwiak, J.; Loska, R.; Mąkosza, M. J. Org. Chem. 2016, 81, 11751. (h) Varaksin, M. V.; Utepova, I. A.; Chupakhin, O. N. Chem. Heterocycl. Compd. 2012, 48, 1213. [Khim. Geterotsikl. Soedin. 2012, 1301.] (i) Varaksin, M. V.; Utepova, I. A.; Chupakhin, O. N.; Charushin, V. N. Tetrahedron 2015, 71, 7077.
(a) Matern, A. I.; Charushin, V. N.; Chupakhin, O. N. Russ. Chem. Rev. 2007, 76, 23. [Usp. Khim. 2007, 76, 27.] (b) Chupakhin, O. N.; Charushin, V. N.; van der Plas, H. C. Tetrahedron 1988, 44, 1. b Berberova, N. T.; Okhlobystin, O. Yu. Chem. Heterocycl. Compd. 1984, 20, 817. [Khim. Geterotsikl. Soedin. 1984, 1011.]
Budyka, M. F.; Terent'ev, P. B.; Kost, A. N. Chem. Heterocycl. Compd. 1978, 14, 663. [Khim. Geterotsikl. Soedin. 1978, 809.]
Borovlev, I. V.; Demidov, O. P.; Saigakova, N. A.; Amangasieva, G. A. Eur. J. Org. Chem. 2014, 7675.
(a) Shchepochkin, A. V.; Chupakhin, O. N.; Charushin, V. N.; Steglenko, D. V.; Minkin, V. I.; Rusinov, G. L.; Matern, A. I. RSC Adv. 2016, 6, 77834. (b) Makhaeva, G. F.; Lushchekina, S. V.; Boltneva, N. P.; Serebryakova, O. G.; Rudakova, E. V.; Ustyugov, A. A.; Bachurin, S. O.; Shchepochkin, A. V.; Chupakhin, O. N.; Charushin, V. N.; Richardson, R. J. Bioorg. Med. Chem. 2017, 25, 5981. (c) Shchepochkin, A. V.; Chupakhin, O. N.; Charushin, V. N.; Rusinov, G. L.; Subbotina, Yu. O.; Slepukhin, P. A.; Budnikova, Yu. G. Russ. Chem. Bull., Int. Ed. 2013, 62, 773. [Izv. Akad. Nauk, Ser. Khim. 2013, 772.]
Gulevskaya, A. V.; Tyaglivaya, I. N.; Verbeeck, S.; Maes, B. U. W.; Tkachuk, A. V. ARKIVOC 2011, (ix), 238.
Garnier, E.; Audoux, J.; Pasquinet, E.; Suzenet, F.; Poullain, D.; Lebret, B.; Guillaumet, G. J. Org. Chem. 2004, 69, 7809.
(a) Bashkin, J. K.; Rains, R.; Stern, M. Green Chem. 1999, 1, G41. (b) Triplett, R. D.; Rains, R. K. US Patent 7504539.
Patriciu, O.-I.; Fînaru, A.-L.; Săndulescu, I.; Guillaumet, G. Synthesis 2007, 3868.
(a) Tondys, H.; van der Plas, H. C.; Wozniak, M. J. Heterocycl. Chem. 1985, 22, 353. (b) Wozniak, M.; Baranski, A.; Nowak, K.; van der Plas, H. C. J. Org. Chem. 1987, 52, 5643.
Demidov, O. P.; Pobedinskaya, D. Yu.; Avakyan, E. K.; Amangasieva, G. A.; Borovlev, I. V. Chem. Heterocycl. Compd. 2018, 54, 875. [Khim. Geterotsikl. Soedin. 2018, 54, 875].
(a) Grzegożek, M. J. Heterocycl. Chem. 2008, 45, 1879. (b) Grzegożek, M.; Szpakiewicz, B.; Kowalski, P. ARKIVOC 2009, (vi), 84.
(a) Stern, M. K.; Cheng, B. K. J. Org. Chem. 1993, 58, 6883. (b) Esser, F.; Pook, K.-H. Synthesis 1992, 596.
Borovlev, I. V.; Demidov, O. P.; Kurnosova, N. A.; Amangasieva, G. A.; Avakyan, E. K. Chem. Heterocycl. Compd. 2015, 51, 170. [Khim. Geterotsikl. Soedin. 2015, 51, 170.]
Demidov, O. P.; Borovlev, I. V.; Amangasieva, G. A.; Avakyan, E. K. Chem. Heterocycl. Compd. 2016, 52, 104. [Khim. Geterotsikl. Soedin. 2016, 52, 104.]
Amangasieva, G. A.; Borovlev, I. V.; Demidov, O. P.; Avakyan, E. K.; Borovleva, A. A. Russ. J. Org. Chem. 2018, 54, 867. [Zh. Org. Khim. 2018, 54, 865.]
(a) Borovlev, I. V.; Demidov, O. P.; Amangasieva, G. A.; Avakyan, E. K.; Borovleva, A. A.; Pobedinskaya, D. Yu. Synthesis 2018, 3520. (b) Avakyan, E. K.; Borovlev, I. V.; Demidov, O. P.; Amangasieva, G. A.; Pobedinskaya, D. Yu. Chem. Heterocycl. Compd. 2017, 53, 1207. [Khim. Geterotsikl. Soedin. 2017, 53, 1207.]
(a) Wróbel, Z.; Kwast, A. Synlett 2007, 1525. (b) Wróbel, Z.; Kwast, A. Synthesis 2010, 3865. (c) Kwast, A.; Stachowska, K.; Trawczyński, A.; Wróbel, Z. Tetrahedron Lett. 2011, 52, 6484. (d) Wróbel, Z.; Stachowska, K.; Grudzień, K.; Kwast, A. Synlett 2011, 1439. (e) Wróbel, Z.; Więcław, M.; Bujok, R.; Wojciechowski, K. Monatsh. Chem. 2013, 144, 1847.
Khan, B.; Khan, A. A.; Bora, D.; Verma, D.; Koley, D. ChemistrySelect 2017, 2, 260.
Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512.
Sharp, J. T.; Gosney, I.; Rowley, A. G. Practical Organic Chemistry; Chapman and Hall: London, 1989.
(a) Zhu, X.; Qiao, L.; Ye, P.; Ying, B.; Xu, J.; Shen, C.; Zhang, P. RSC Adv. 2016, 6, 89979. (b) He, Y.; Zhao, N.; Qiu, L.; Zhang, X.; Fan, X. Org. Lett. 2016, 18, 6054. (c) Mondal, S.; Samanta, S.; Hajra, A. Adv. Synth. Catal. 2018, 360, 1026. (d) Whiteoak, C. J.; Planas, O.; Company, A.; Ribas, X. Adv. Synth. Catal. 2016, 358, 1679. (e) Wang, Y.; Yu, F.; Han, X.; Li, M.; Tong, Y.; Ding, J.; Hou, H. Inorg. Chem. 2017, 56, 5953.
Ma̧kosza, M.; Białecki, M. J. Org. Chem. 1998, 63, 4878.
CrysAlisPro, version 1.171.38.41; Rigaku Oxford Diffraction, 2015. https://www.rigaku.com/en/products/smc/crysalis.
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3.
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3.
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2019, 55(7), 623–631
Electronic supplementary material
ESM 1
(PDF 1726 kb)
Rights and permissions
About this article

Cite this article
Amangasieva, G.А., Avakyan, E.K., Demidov, O.P. et al. SNH Amidation of nitroquinolines: synthesis of amides on the basis of nitro- and nitrosoquinolines. Chem Heterocycl Comp 55, 623–631 (2019). https://doi.org/10.1007/s10593-019-02508-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-019-02508-3