

An effective method has been developed for the synthesis of cyclic propargylamines by aminomethylation of α,ω-diacetylenes with N-alkyl-substituted 1,5,3-dioxazepanes in the presence of copper-containing catalysts.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Azacycloalkynes have been previously obtained in reactions of primary amines with α,ω-dihaloalkadiynes,1 by intramolecular cyclization of diacetylenic gem-amino ethers under high dilution conditions,2 by a three-component cyclocondensation of terminal alkadiynes, aldehydes, and amines (A3-coupling) in the presence of СuCl catalyst,3 as well as by aminomethylation of α,ω-diacetylenes by using N,N-bis(ethoxymethyl)amines in the presence of СuBr2 catalyst.4
It has been previously reported that 1,5,3-dioxazepanes5 can be effectively used as Mannich bases in aminomethylation reactions of β-keto esters6 and cyclopentanone.7 In the current work, we present data about the synthesis of azacycloalkadiynes by catalytic cycloaminomethylation of α,ω-diacetylenes using N-substituted 1,5,3-dioxazepanes.
The interest toward azacycloalkadiynes is motivated by their applications in organic synthesis, for example, in thermal intramolecular cyclization reactions of 1,6-diazacyclodeca-3,8-diynes8 or 1-thia-6-azacyclodeca-3,8-diynes.9
While continuing our exploration of aminomethylation reactions of terminal acetylenes using 1,5,3-dioxazepanes,10 as well as for the purpose of developing an effective method for the synthesis of azacyclodiynes, we studied the reaction of α,ω-diacetylenes with N-substituted 1,5,3-dioxazepanes 1a–с in the presence of copper-containing catalysts. The selection of catalysts on the basis of copper compounds was justified by their high activity in reactions between terminal acetylenes and N,N-bis(ethoxymethyl)amines.4
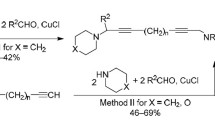
By using the model reaction of 3-butyl-1,5,3-dioxazepane (1b) with 1,7-octadiyne (2b), we screened catalysts that were either pure copper compounds (СuCl (5% yield of the product), CuBr (16%), CuI (25%), CuCl2 (36%), CuBr2 (65%), CuSO4 (7%)) or immobilized copper catalysts on solid supports (CuCl2·2Н2О–γ-Al2O3 (25% yield of the product), CuBr2–γ-Al2O3 (23%), CuBr2–microSiO2 (7%), CuBr2–mesoSiO2 (16%), CuBr2–macroSiO2 (15%), CuBr2–ASM-40 (14%), CuBr2–НY-BS (10%)). The highest yield (~65%) of 1-butyl-1-azacycloundeca-3,9-diyne (3d) was achieved by using 5 mol % of СuBr2 catalyst at 80°С over 6 h in toluene under argon atmosphere. The reaction of α,ω-alkadiynes 2а–d with 1,5,3-dioxazepanes 1a–с under these conditions led to 1-alkyl-1-azacycloalkadiynes 3а–g in 42–72% yields (Scheme 1). The spectral characteristics of compounds 3c,d,e,g matched the literature data.4

Scheme 1.
Cycloalkyl(cyclopropyl,cyclopentyl,cyclohexyl)- and aryl-substituted 1,5,3-dioxazepanes did not react with α,ω-diacetylenes under the aforementioned conditions, because the indicated 1,5,3-dioxazepanes were converted to 1,3,5-triazines under the reaction conditions developed during our study. The α,ω-diacetylenes containing a heteroatom – (di(propyn-2-yl)ether and N,N-di(propyn-2-yl)amine) showed low activity in the reaction with alkyl-substituted 1,5,3-dioxazepanes, probably due to conformational rigidity caused by the effects of lone electron pairs of the heteroatom on the structure of α,ω-diacetylene.
Thus, we have developed a new one-pot method for the synthesis of azacyclodiynes by cycloaminomethylation of α,ω-diacetylenes using N-substituted 1,5,3-dioxazepanes in the presence of СuBr2 catalyst.
Experimental
IR spectra were recorded on a Bruker Vertex 70v spectrometer for samples in the form of thin films. 1Н and 13С NMR spectra were recorded on Bruker Avance 400 (400 and 100 MHz, respectively) and Bruker Ascend 500 (500 and 125 MHz, respectively) spectrometers in CDCl3. Homonuclear (COSY) and heteronuclear (1Н–13С HSQC, 1Н–13С HMBC) two-dimensional NMR experiments for compounds 3а–e were performed on a Bruker Avance 500 spectrometer. The chemical shifts were determined relative to solvent signals (δН 7.28 ppm and δС 77.1 ppm, respectively). GC/MS analyses were performed on a Shimadzu GC 2010 chromatograph with a Shimadzu GCMS-QP2010 Ultra mass selective detector. The gas chromatograph was equipped with a Supelco 5ms capillary column (60 m × 0.25 mm × 0.25 μm), carrier gas – helium, injector temperature 260°С, interface temperature 260°С, ion source temperature 200°С, EI ionization (70 eV). The content of С, Н, and N was determined with a Carlo Erba 1106 СHN-analyzer. Gas chromatography was performed on a Shimadzu GC-9A with a flame ionization detector, using SE-30 stationary phase (5%) on a Chromaton N-AW-HMDS support, 2000 × 3 mm packed steel column, temperature program 50–280°С, 8°C/min, the carrier gas was helium. Thin-layer chromatography was performed on Sorbfil PTSKh-AF-A plates, with visualization in iodine vapor. Individual compounds were isolated by chromatographic separation on KSK silica gel (50–160 μm). The eluent used for column chromatography is indicated for each compound.
The starting α,ω-diacetylenes 2а–d with ≥98% assay were commercially available from Acros Organics and were used without additional purification, while 1,5,3-dioxazepanes 1a–с were synthesized according to published procedures.5 Copper salts (CuBr, CuI, CuCl2, CuBr2, CuCl2·2Н2О, CuSO4, and Cu(CH3CO2)2) with ≥99% assay were commercially available from Acros Organics. The catalysts on solid support were obtained by coating copper salts on solid carriers. The procedure involved treatment of γ-Al2O3, micro-meso, meso, and macroporous silica gels, as well as acidic amorphous alumosilicate and micro-mesomacroporous zeolite HY (HY-BS) with the aforementioned copper salts as solutions in alcohol or ether, followed by drying at 110°С according to a published method.11 All samples contained 10–12 mass % of the respective copper salts.
Synthesis of 1-alkyl-1-azacycloalkadiynes 3а–g (General method). A Schlenk vessel was charged under argon atmosphere with the appropriate 1,5,3-dioxazepane 1a–с (1 mmol), toluene (3 ml), α,ω-diacetylene 2а–d (1 mmol), and СuBr2 (0.01 g, 0.05 mmol). The reaction mixture was stirred at 80°С for 6 h and filtered through a layer of silica gel; the solvent was evaporated on a rotary evaporator. The product was isolated by column chromatography.
1-Propyl-1-azacyclododeca-3,10-diyne (3a). Yield 0.085 g (42%), yellow oil. Rf 0.53 (hexane–dichloromethane, 1:1). IR spectrum, ν, cm–1: 1140 (C–N), 1432 (CH3), 1457 (CH2), 2258 (С≡C), 2860 (CH2), 2934 (СН3). 1H NMR spectrum (500 MHz), δ, ppm (J, Hz): 0.93 (3H, t, J = 7.5, CH3); 1.48–1.53 (8H, m, (C≡СCH2СН2)2СН2, CH2CH3); 2.21–2.23 (4H, m, N(CH2С≡СCH2)2); 2.45 (2Н, t, J = 7.5, CH3CH2CH2N); 3.38 (4Н, s, N(CH2C≡C)2). 13C NMR spectrum (125 MHz), δ, ppm: 12.0 (CH3); 18.7 (2C≡CCH2); 20.7 (CH2СН3); 28.3 ((C≡CCH2CH2)2СН2); 28.5 (2С≡СCH2CH2); 42.5 (N(CH2C≡C)2); 54.9 (CH2CH2N); 75.1 (N(CH2C≡C)2); 84.8 (N(CH2C≡C)2). Mass spectrum, m/z (Irel, %): 203 [М]+ (11), 188 [М–CH3]+ (3), 174 [M–CН2СН3]+ (100), 160 [М–CH3(CH2)2]+ (9), 132 [M–N(CH2)3CH3]+ (7). Found, %: С 82.76; Н 10.34; N 6.91. С14Н21N. Calculated, %: С 82.70; Н 10.41; N 6.89.
1-Propyl-1-azacyclotrideca-3,11-diyne (3b). Yield 0.132 g (60%), yellow oil. Rf 0.45 (hexane–dichloromethane, 1:1). IR spectrum, ν, cm–1: 1170 (C–N), 1433 (CH3), 1461 (CH2), 2118 (С≡C), 2859 (CH2), 2933 (СН3). 1H NMR spectrum (500 MHz), δ, ppm (J, Hz): 0.93 (3H, t, J = 7.5, CH3); 1.38–1.44 (4H, m, (C≡ССН2CH2СН2)2); 1.49–1.56 (6H, m, (C≡CСН2CH2CH2)2, CH2СН2N); 2.19–2.22 (4H, m, N(СН2С≡СCH2)2); 2.47 (2Н, t, J = 7.5, CH2CH2N); 3.39 (4Н, s, N(CH2C≡C)2). 13C NMR spectrum (125 MHz), δ, ppm: 11.96 (CH3); 18.7 (2C≡CCH2); 20.7 (CH2СН3); 28.4 (2C≡CCH2CH2СН2); 28.8 (2С≡СCH2CH2); 42.5 (N(CH2C≡C)2); 54.9 (CH2CH2N); 75.1 (N(CH2C≡C)2); 84.9 (N(CH2C≡C)2). Mass spectrum, m/z (Irel, %): 217 [М]+ (10), 202 [М–CH3]+ (3), 188 [M–CН2СН3]+ (100), 174 [М–CH3(CH2)2]+ (3), 160 [M–N(CH2)3CH3]+ (3), 91 [NH(CH2C≡C)2]+ (18). Found, %: С 82.94; Н 10.61; N 6.45. С15Н23N. Calculated, %: С 82.89; Н 10.67; N 6.44.
1-Butyl-3,4,8,9-tetradehydro-1,2,5,6,7,10-hexahydroazecine (3c). Yield 0.085 g (45%), yellow oil. Rf 0.53 (cyclohexane–ethyl acetate–CН2Сl2, 1:10:2). IR spectrum, ν, cm–1: 1110 (C–N), 1432 (CH3), 1455 (CH2), 2220 (С≡C), 2861 (CH2), 2931 (СН3). 1H NMR spectrum (500 MHz), δ, ppm (J, Hz): 0.94 (3H, t, J = 9.0, CH3); 1.33–1.39 (2H, m, CH2СН3); 1.40–1.49 (2H, m, CH2CH2СН3); 1.71–1.76 (2H, m, (C≡CCH2)2CH2); 2.32–2.35 (4H, m, (С≡СCH2)2CH2); 2.50 (2Н, t, J = 9.0, CH2CH2N); 3.38 (4Н, s, N(CH2C≡C)2). 13C NMR spectrum (125 MHz), δ, ppm: 14.0 (CH3); 18.0 ((C≡CCH2)2CH2); 20.6 (CH2СН3); 28.2 ((С≡СCH2)2CH2); 29.6 (CH2CH2СН3); 42.6 (N(CH2C≡C)2); 52.7 (CH2CH2N); 75.6 (N(CH2C≡C)2); 84.0 (N(CH2C≡C)2). Mass spectrum, m/z (Irel, %): 188 [M–Н]+ (6), 174 [M–CН3]+ (6), 146 [М–(CH2)2CH3]+ (100), 117 [M–NH(CH2)3CH3]+ (45), 103 [М–CH3(CH2)3NHCH2]+ (13), 91 [NH(CH2C≡C)2]+ (83). Found, %: С 82.53; Н 10.07; N 7.35. С13Н19N. Calculated, %: С 82.48; Н 10.12; N 7.40.
1-Butyl-1-azacycloundeca-3,9-diyne (3d). Yield 0.132 g (65%), yellow oil. Rf 0.60 (hexane–ethyl acetate, 1:2). IR spectrum, ν, cm–1: 1111 (C–N), 1325 (C–N), 1385 (CH3), 1432 (CH3), 1458 (CH2), 2230 (C≡C), 2863 (CH2), 2931 (СН3). 1H NMR spectrum (400 MHz), δ, ppm (J, Hz): 0.94 (3H, t, J = 7.2, CH3); 1.33–1.38 (2H, m, CH2СН3); 1.40–1.48 (2H, m, CH2CH2CH3); 1.63–1.65 (4H, m, (С≡СCH2CH2)2); 2.24 (4Н, br. s, (C≡CCH2CH2)2); 2.50 (2H, t, J = 7.2, CH2CH2N); 3.38 (4Н, s, N(CH2C≡C)2). 13C NMR spectrum (100 MHz), δ, ppm: 14.0 (CH3); 18.2 (2C≡CCH2CH2); 20.6 (CH2СН3); 27.9 (2С≡СCH2CH2); 29.6 (CH2CH2СН3); 42.5 (N(CH2C≡C)2); 52.6 (CH2CH2N); 75.3 (N(CH2C≡C)2); 84.4 (N(CH2C≡C)2). Mass spectrum, m/z (Irel, %): 202 [M–Н]+ (9), 160 [M–(CH2)2CН3]+ (100), 117 [M–CH2NH(CH2)3CH3]+ (38), 91 [М–N(CH2C≡C)2]+ (51). Found, %: С 82.74; Н 10.45; N 6.92. C14H21N. Calculated, %: C 82.70; H 10.41; C 6.89.
1-Butyl-1-azacyclotrideca-3,11-diyne (3e). Yield 0.167 g (72%), yellow oil. Rf 0.45 (cyclohexane–ethyl acetate–CН2Сl2, 1:10:2). IR spectrum, ν, cm–1: 1111, 1144 (C–N), 1361 (CH3), 1436 (CH2), 2261 (C≡C), 2857 (СН2), 2930 (CH3). 1H NMR spectrum (500 MHz), δ, ppm (J, Hz): 0.94 (3H, t, J = 7.2 CH3); 1.35–1.38 (2H, m, CH2СН3); 1.39–1.43 (4H, m, (С≡СCH2CH2CH2)2); 1.44–1.49 (2H, m, CH2CH2СН3); 1.51–1.53 (4H, m, (С≡СCH2CH2СН2)2); 2.21 (4Н, t, J = 7.0 (С≡СCH2CH2СН2)2); 2.50 (2Н, t, J = 7.5, CH2CH2N); 3.38 (4Н, s, N(CH2C≡C)2). 13C NMR spectrum (125 MHz), δ, ppm: 14.0 (CH3); 18.7 (2C≡CCH2CH2CH2); 20.7 (CH2СН3); 28.4 (2С≡СCH2CH2СН2); 28.8 (2С≡СCH2CH2СН2); 29.6 (CH2CH2СН3); 42.6 (N(CH2C≡C)2); 52.6 (CH2CH2N); 75.1 (N(CH2C≡C)2); 84.9 (N(CH2C≡C)2). Mass spectrum, m/z (Irel, %): 231 [M]+ (6), 207 [M–C≡С]+ (27), 188 [M–(CH2)2CH3]+ (87), 40 [CNCH2]+ (100). Found, %: С 83.11; Н 10.92; N 6.01. С16Н25N. Calculated, %: С 83.06; Н 10.89; N 6.05.
1- tert -Butyl-3,4,8,9-tetradehydro-1,2,5,6,7,10-hexahydroazecine (3f). Yield 0.12 g (63%), yellow oil. Rf 0.38 (hexane–ethyl acetate–CН2Сl2, 1:2:1). IR spectrum, ν, cm–1: 1018 (C–N), 1366 (СН3), 1392 (СН3), 1453 (CH3), 2118 (С≡С), 2200 (С≡С), 2910 (CH2), 2960 (СН3). 1H NMR spectrum (500 MHz), δ, ppm (J, Hz): 1.20 (9H, s, 3CH3); 1.68–1.76 (2H, m, (C≡CСН2)2CH2); 2.29–2.36 (4H, m, (С≡СCH2)2); 3.60 (4Н, s, N(CH2C≡C)2). 13C NMR spectrum (125 MHz), δ, ppm: 18.2 ((2C≡CCH2)2); 27.5 ((СН3)3); 28.0 ((С≡СCH2)2CH2); 36.8 (N(CH2C≡C)2); 55.0 (C(СН3)3); 78.1 (N(CH2C≡C)2); 83.3 (N(CH2C≡C)2). Mass spectrum, m/z (Irel, %): 189 [M]+ (9), 174 [M–CН3]+ (100), 144 [М–3(CH3)]+ (13), 132 [М–C(CH3)]+ (30), 117 [M–NHC(CH3)3]+ (40), 91 [NH(CH2C≡C)2]+ (45). Found, %: С 82.61; Н 10.08; N 7.43. С13Н19N. Calculated, %: С 82.48; Н 10.12; N 7.40.
1-( tert- Butyl)-1-azacycloundeca-3,9-diyne (3g). Yield 0.103 g (51%), yellow oil. Rf 0.48 (hexane–ethyl acetate–CН2Сl2, 1:2:1). IR spectrum, ν, cm–1: 1202 (С(СН3)3), 1262 (C–N), 1364 (CH3), 1390 (CH3), 1431 (CH3), 1457 (CH2), 2130 (C≡С), 2863 (CH2), 2938 (CH3). 1H NMR spectrum (400 MHz), δ, ppm (J, Hz): 1.17 (9H, s, 3CH3); 1.59–1.62 (4H, m, (CH2CH2)2); 2.20 (4H, br. s, (CH2CH2)2); 3.57 (4H, s, N(CH2C≡C)2). 13C NMR spectrum (100 MHz), δ, ppm: 18.5 (2C≡CCH2CH2); 27.5 (СН3); 27.9 ((С≡СCH2СН2)2); 36.7 (N(CH2)2); 54.9 (NC(CH3)3); 77.8 (N(CH2C≡C)2); 83.7 (N(CH2C≡C)2). Mass spectrum, m/z (Irel, %): 203 [M]+ (7), 188 [M–СН3]+ (100), 146 [М–C(CH3)3]+ (9), 117 [M–СН2NНС(CH3)3]+ (16), 91 [NН(CH2C≡C)2]+ (29). Found, %: С 82.83; Н 10.52; N 6.84. С14Н21N. Calculated, %: С 82.70; Н 10.41; N 6.89.
References
(a) Gleiter, R.; Ritter, J. Liebigs Ann./Recl. 1997, 2113. (b) Gleiter, R.; Ritter, J.; Irngartinger, H.; Lichtenthäler, J. Tetrahedron Lett. 1991, 32, 2887.
Epsztein, R.; Goff Le, N. Tetrahedron Lett. 1985, 26, 3203.
(a) Pang, T.; Yang, Q.; Gao, M.; Wang, M.; Wu, A. Synlett 2011, 3046. (b) Cai, Q.; Yang, Q.-W.; Zhang, J.-M. Chin. J. Struct. Chem. 2014, 33, 785.
Khabibullina, G. R.; Zaynullina, F. T.; Valiakhmetova, А. R.; Ibragimov, A. G.; Dzhemilev, U. M. Synthesis 2016, 2294.
Kapnang, H.; Charles, G. Tetrahedron Lett. 1983, 24, 1597.
Sparrow, K.; Barker, D.; Brimble, M. A. Tetrahedron 2012, 68, 1017.
Ooka, K.; Inoue, H. JP Patent 2008094744.
Gleiter, R.; Ritter, J. Angew. Chem., Int. Ed. 1994, 33, 2470.
Gleiter, R.; Ritter, J.; Irngartinger, H.; Lichtenthäler, J. Tetrahedron Lett. 1991, 32, 2883.
Khabibullina, G. R.; Zaynullina, F. T.; Karamzina, D. S.; Ibragimov, A. G.; Dzhemilev, U. M. Tetrahedron 2017 , 73, 2367.
Veklov, V. A.; Kutepov, B. I.; Talipova, R. R.; Grigor'eva, N. G.; Dzhemilev, U. M.; Drozdov, V. A. RU Patent 2420455; Byul. Izobret. 2011, (16).
This work was supported by grant of the President of the Russian Federation (NSh-6651.2016.3) and the Russian Science Foundation (RSF-DST No.16-43-02010).
Structural characterization of the compounds was performed at the Center for Collective Use “Agidel” at the Institute of Petrochemistry and Catalysis of the Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
A Supplementary information file containing 1Н and 13С MR spectra of compounds 3b and 3e is available at the journal website at http://springerlink.bibliotecabuap.elogim.com/journal/10593.
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2018, 54(1), 86–88
Electronic supplementary material
ESM 1
(PDF 263 kb)
Rights and permissions
About this article

Cite this article
Khabibullina, G.R., Zaynullina, F.T., Kutepov, B.I. et al. One-pot synthesis of azacyclodiynes by reaction of α,ω-diacetylenes with 1,5,3-dioxazepanes using copper-containing catalysts. Chem Heterocycl Comp 54, 86–88 (2018). https://doi.org/10.1007/s10593-018-2235-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-018-2235-9