

Methyl 3-(4-methylfurazan-3-yl)-1Н-pyrazole-5-carboxylate was obtained by condensation of 3-acetyl-4-methylfurazan with diethyl oxalate with subsequent treatment of the β-diketoester intermediate with hydrazine, and was nitrated to produce 3(5)-(3-methylfurazan-4-yl)-4-nitro-1Н-pyrazole-5(3)-carboxylic acid. The amide of this acid was used in a Hofmann rearrangement, providing 3-amino-5-(4-methylfurazan-3-yl)-4-nitro-1Н-pyrazole, nitration of which yielded the respective nitramine.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Nitropyrazoles containing various functional groups are commonly used in targeted synthesis of pyrazole-containing compounds for a range of applications.1 Of significant interest are nitropyrazoles containing an additional carboxy functionality. Thus, nitropyrazolecarboxylic acids (NPCA) and their derivatives serve as key precursors for the synthesis of pharmacologically relevant purine analogs: pyrazolo[3,4-d]pyrimidines,2 pyrazolo[4,3-d]-pyrimidines,3 pyrazolo[1,5-a]pyrimidines,4 and pyrazolo-[4,3-d]triazinones.5 Some of the compounds obtained on the basis of NPCA have been identified as selective inhibitors of such enzymes as phosphodiesterase (PDE 5/6/9), SCD1, HIF-1,3 , 5 , 6 cannabinoid and nicotinic receptors,7 inhibitors of protein aggregation,8 calcium channel blockers,9 and pesticides.9 , 10 Certain members of the NPCA family have been recently used for the synthesis of aminonitropyrazoles11,12, – 13 with potential applications as low sensitivity energetic compounds.14
A review of the literature shows that none of the many reported examples of NPCA derivatives contains another heterocycle as a substituent in the pyrazole ring. One of the directions recently explored by our group is the synthesis and reactivity studies of С- and N-(hetaryl)nitropyrazoles consisting of a nitrogen-rich heterocycle linked to a nitropyrazole ring,15,16,17, – 18 particularly furazanylnitro-pyrazoles.15 , 16 In a continuation of this research effort, the current work is focused on the synthesis and transformations of 3(5)-(3-methylfurazan-4-yl)-4-nitro-1Н-pyrazole-5(3)-carboxylic acid (1), representing the first example of hetarylnitro-pyrazolecarboxylic acids.
The traditional methods for the synthesis of NPCA derivatives are acidic nitration of pyrazolecarboxylic acids12 , 19 and oxidation of methyl group in С-methylnitropyrazoles.11 , 20,21,22, – 23 However, there are no published procedures available for the synthesis of the necessary methylnitropyrazoles, as well as pyrazolecarboxylic acids and their derivatives.
The most common approach to the synthesis of 3(5)-substituted pyrazolecarboxylic acids is based on the interaction of various hydrazines with synthetic equivalents of acylpyruvates (mostly esters), which in turn can be obtained by Claisen condensation of the respective methyl ketones with dialkyl oxalates.24 This approach was also found to be effective in our case.
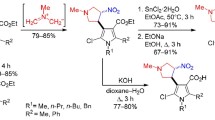
Indeed, condensation of the available 3-acetyl-4-methylfurazan25 (2) with diethyl oxalate in the presence of sodium methoxide allowed to prepare the respective pyruvate 3 in a high yield (Scheme 1). The heterocyclization reaction of β-diketoester 3 proceeded smoothly upon treatment with hydrazine, with the formation of pyrazolecarboxylic acid ester 4. The subsequent alkaline hydrolysis of this ester gave the target carboxylic acid 5. The overall yield of acid 5 over the three steps shown in Scheme 1 reached 67%.

Scheme 1.
We also demonstrated that acid 5 can be obtained by an alternative method, namely, by direct carboxylation of the pyrazole ring.26 The synthesis of pyrazolecarboxylic acids by this method is a specific example of a more general method for the functionalization of pyrazole ring, based on the interaction between С-anions derived from N-substituted pyrazoles with electrophiles.27 The starting material selected for synthesis of carboxylic acid 5 according to this method was our previously obtained N-unsubstituted furazanylpyrazole 6.15
It is well known that N-unsubstituted arylpyrazoles are NH acids (рK a 13–14),28 therefore the С-deprotonation of pyrazole ring in compound 6 can be achieved only by preventing the formation of an N-anion by introducing an easily removable protecting group at the ring nitrogen atom. For this reason, an acid-catalyzed addition of ethyl vinyl ether to pyrazole 6 was used for the preparation of N-substituted pyrazole 7 as a mixture of 1,3- and 1,5-isomers 7а and 7b in 9:1 ratio (Scheme 2). This mixture of isomers was used in the further reaction without separation. The С-deprotonation of pyrazoles 7а and 7b in the presence of n-BuLi proceeded at the position that was adjacent to the ring nitrogen atom. Carboxylation of the obtained anions 7a',b' by treatment with СО2 gave the respective carbanions 7a",b". The subsequent acidcatalyzed removal of protecting group allowed to obtain the N-unsubstituted furazanylpyrazolecarboxylic acid 5 with the overall yield of 64% (Scheme 2).

Scheme 2.
It should be noted that in the case of the N-unsubstituted pyrazole 6 the treatment with a twofold excess of n-BuLi and then СО2 led to a completely different result. The first equivalent of n-BuLi transformed compound 6 to its N-anion 6', thus preventing С-deprotonation of the pyrazole ring. As shown in Scheme 3, in this case the addition of a second equivalent of n-BuLi resulted in С-deprotonation of the methyl group at the furazan ring, which is typical for methylfurazans.29 The obtained carbanion 6” was converted by treatment with СО2 to the furazanylacetate anion 6”', which gave the furazanylacetic acid 8 after treatment with aqueous hydrochloric acid.

Scheme 3.
Thus, two fundamentally different methods for the synthesis of acid 5 have been developed. However, it should be noted that the method based on using acetyl-methylfurazan 2 as precursor was more convenient, since it did not require anhydrous solvents and low temperatures.
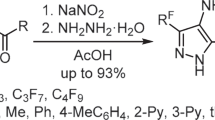
We have shown that, although the nitration of acid 5 with a mixture of sulfuric and nitric acids resulted in the introduction of a nitro group at position 4, which was the most reactive site toward electrophiles,1 , 30 there was also a simultaneous decarboxylation at the pyrazole ring position 3. This process resulted in the formation of 3(5)-furazanyl-4-nitropyrazole 9 (Scheme 4), which had been previously obtained by nitration of 3(5)-furazanylpyrazole 6.15 Decarboxylation reactions of pyrazolecarboxylic acids in nitrating mixtures of sulfuric and nitric acids have been previously reported in the literature. However, in contrast to the reaction performed in our study, the previously described processes were always accompanied by ipsonitration.12 , 19 b, 22 , 31 , 32

Scheme 4.
We established that the synthesis of nitropyrazolecarboxylic acid 1 can be achieved by nitration of its methyl ester 4. The ester group apparently played the role of protecting group during nitration in strongly acidic media and was removed by acid-catalyzed hydrolysis after dilution with water during the workup of reaction mixture (Scheme 5).

Scheme 5.
A characteristic feature of pyrazole reactivity is their ability to form stable nitro derivatives at the ring nitrogen atom. N-Nitropyrazoles not only are common intermediates in synthetic processes involving pyrazole derivatives,1 , 30 but also have been characterized as exogenous nitric oxide donors.33 For example, it was demonstrated with in vivo experiments that, due to their ability to release nitric oxide in the body, N-nitropyrazoles facilitate the regeneration of retina after ischemic insult, with the strongest activity shown by derivatives of pyrazolecarboxylic acids.34
We found that the N-nitration of ester 4 with acetyl nitrate resulted in the formation of two isomeric N-nitro derivatives 10а and 10b in approximately 1:1 ratio according to the data of 1Н and 13С spectroscopy (Scheme 5). However, we were not able to separate these isomers, as they had low stability under the chromatographic conditions and decomposed during crystallization attempts. It should be noted that the formation of a mixture of N-nitro isomers was previously reported only for the N-nitration reaction of 3(5)-methylpyrazole.35 Apparently, the mixture of isomers in the case of methyl ester 4 was created due to the presence of two 3(5)-substituents with comparable electron-withdrawing strength.
In accordance with Scheme 6, the carboxyl group of compound 1 was converted to an amino group. The key step on this route was a Hofmann rearrangement, which is effective for the synthesis of aminonitropyrazoles.11 , 13 , 21 Methyl ester 11, obtained from acid 1 and MeOH in the presence of SOCl2, was treated with aqueous ammonia. The reaction was performed at 40°C with the addition of MeOH for improving the solubility of ester 11. The treatment of amide 12 with NaBrO in aqueous medium resulted in Hofmann rearrangement, leading to the formation of aminonitropyrazole 13 in a high yield.

Scheme 6.
It is well known that nitramino derivatives of azoles are of interest as energetic compounds.36,37, – 38 For this reason, we studied the possibility of N-nitration in the case of aminopyrazole 13, which has two possible sites of nitration – the amino group and the ring NH moiety. The reaction was performed by using acetyl nitrate under the same conditions as those of our earlier synthesis of N-nitropyrazoles with furazanyl substituent.15 It was found that, similarly to the case of monocyclic aminopyrazoles,11 , 18 , 36 the presence of a С-amino group in the nitropyrazole 13 completely changed the direction of the nitration reaction. The reaction proceeded exclusively at the amino group, without affecting the NH moiety of pyrazole ring, and provided the nitramine 14 in a high yield.
Thus, the traditional methods that have been used in the chemistry of monocyclic nitropyrazoles were adapted for the synthesis of 3(5)-(3-methylfurazan-4-yl)-4-nitro-1Н-pyrazole-5(3)-carboxylic acid – the first representative of С-hetaryl-substituted pyrazoles simultaneously containing two functional groups – СО2Н and NO2. It was shown that the presence of a hetaryl substituent in the pyrazole ring had some specific effects on the reactivity, but did not prevent the introduction of the desired functional groups. The obtained data can be useful for planning the synthesis of other structurally related compounds.
Experimental
IR spectra were recorded on a Bruker Alpha instrument in KBr pellets. 1Н, 13C, and 14N NMR spectra were acquired on a Bruker AМ-300 instrument (300, 75, and 21 MHz, respectively) in DMSO-d 6 (unless indicated otherwise) at 299 K. The chemical shifts for 1H and 13C nuclei are reported relative to TMS, for 14N nuclei – relative to MeNO2. Mass spectra were recorded on a Finnigan MAT Incos 50 instrument (direct introduction of samples, EI ionization at 70 eV). High-resolution mass spectra with electrospray ionization were recorded on a Bruker MicroOTOF II instrument. Elemental analysis was performed on a PerkinElmer 2400 Series II instrument. Melting points were determined by Kofler method on a Boetius hot stage (heating rate 4°C/min) and were not corrected. The reaction progress and purity of the obtained compounds were controlled by TLC on Merck Silicagel 60 F254 plates.
The starting materials – 3-acetyl-4-methylfurazan (2)25 and 3-(4-methylfurazan-3-yl)pyrazole (6)15 were obtained according to published procedures.
5-(4-Methyl-1,2,5-oxadiazol-3-yl)-4-nitro-1 Н -pyrazole-3-carboxylic acid (1). A solution of ester 4 (0.25 g, 1.2 mmol) in HNO3 (2.5 ml, ρ 1.50 g/cm3) was stirred for 1.5 h at 80–85°C. The reaction mixture was cooled, poured into ice water (10 ml), extracted with Et2O (3×15 ml), and the organic layer was dried over anhydrous Na2SO4. The solvent was removed by evaporation at reduced pressure, and a milky-white solid residue was obtained. Yield 0.26 g (89%), white, needle-shaped crystals, mp 203–204°C (CHCl3). IR spectrum, ν, cm−1: 3268 (s), 2911 (w), 1724 (s), 1533 (s), 1448 (m), 1383 (m), 1302 (m), 1238 (m), 1215 (s), 979 (m), 910 (w), 833 (w), 768 (m). 1H NMR spectrum, δ, ppm: 15.37 (2Н, br. s, NH, ОН); 2.54 (3Н, s, CH3). 13C NMR spectrum, δ, ppm: 157.9 (С=О); 151.3 (С furazan); 145.9 (С furazan); 133.0; 132.6 (br. s, С-4); 131.6; 8.9 (CH3). 14N NMR spectrum, δ, ppm: −20.67. Mass spectrum, m/z: 239 [M]+, 221 [M−H2O]+. Found, %: C 34.67; H 2.04; N 28.84. С7H5N5O5. Calculated, %: C 35.16; H 2.11; N 29.28.
Methyl 2-hydroxy-4-(4-methyl-1,2,5-oxadiazol-3-yl)-4-oxabut-2-enecarboxylate (3). A solution of acetylfurazan 2 (1.0 g, 7.9 mmol) and diethyl oxalate (1.4 ml, 10.3 mmol) in MeOH (10 ml) was stirred at room temperature and treated by dropwise addition of MeONa solution, which was separately prepared from sodium metal (0.2 g, 8.7 mmol) and MeOH (15 ml). The reaction mixture after 48 h was poured into Et2O (20 ml); the precipitate that formed was filtered off, washed with Et2O, and air-dried. The obtained sodium salt of compound 3 was suspended in water (50 ml), acidified with HCl to pH 1, the obtained light cream-colored precipitate was filtered off, washed with water, and air-dried. Yield 1.50 g (89%). The product was characterized without additional purification. IR spectrum, ν, cm−1: 3095 (w), 1746 (s), 1632 (s), 1481 (m), 1438 (m), 1379 (m), 1312 (s), 1275 (s), 1245 (s), 1120 (m), 1035 (w), 992 (w), 973 (m), 831 (m), 786 (m), 765 (w), 681 (m). 1H NMR spectrum, δ, ppm: 6.82 (1Н, s, СH); 3.84 (3Н, s, ОСН3); 2.52 (3Н, s, CH3). 1H NMR spectrum (CDCl3) δ, ppm: 13.72 (1Н, br. s, ОН); 7.30 (1Н, s, СH); 3.96 (3Н, s, ОСН3); 2.64 (3Н, s, CH3). 13C NMR spectrum (CDCl3), δ, ppm: 184.8 (С=О); 166.0; 161.6; 151.3 (С furazan); 150.8 (С furazan); 101.8 (CH); 53.5 (OCH3); 9.4 (CH3).
Methyl 5-(4-methyl-1,2,5-oxadiazol-3-yl)-1 Н -pyrazole-3-carboxylate (4). A solution of furazan 3 (1.5 g, 7.1 mmol) in AcOH (8 ml) was treated by dropwise addition of N2H4·H2O (0.4 ml, 8.0 mmol) and stirred at room temperature for 1 h. The reaction mixture was poured into water (5 ml), the precipitate that formed was filtered off, washed with water, and air-dried. The filtrate was extracted with CH2Cl2 (2×15 ml), then the organic layer was dried over anhydrous Na2SO4, the solvent was removed by evaporation at reduced pressure, giving a solid residue. Both precipitates were combined and crystallized from CHCl3. Yield 1.27 g (86%), colorless platelets, mp 153–154°C. IR spectrum, ν, cm−1: 3228 (s), 1733 (s), 1433 (w), 1388 (w), 1285 (m), 1245 (s), 1207 (w), 1151 (w), 1036 (w), 1015 (w), 956 (w), 895 (w), 847 (w), 780 (w). 1H NMR spectrum, δ, ppm: 14.70 (1H, br. s, NH); 7.32 (1H, s, H-4); 3.89 (3H, s, OCH3); 2.61 (3H, s, CH3). 1H NMR spectrum (CDCl3), δ, ppm: 10.67 (1H, br. s, NH); 7.40 (1H, s, H-4); 4.00 (3H, s, ОCH3); 2.69 (3H, s, CH3). 13C NMR spectrum, δ, ppm: 159.2 (C=O); 150.3 (С furazan); 147.8 (С furazan); 137.8; 136.1; 108.4; 52.1 (OCH3); 9.2 (CH3). 13C NMR spectrum (CDCl3), δ, ppm: 159.9 (C=O); 150.1 (С furazan); 147.9 (С furazan); 139.7; 136.1; 108.8; 52.4 (OCH3); 9.8 (CH3). Found, m/z: 209.0675 [M+H]+. C8H9N4O3. Calculated, m/z: 209.0674. Found, %: C 46.25; H 3.69; N 26.88. С8H8N4O3. Calculated, %: C 46.16; H 3.87; N 26.91.
5-(4-Methyl-1,2,5-oxadiazol-3-yl)-1 Н -pyrazole-3-carboxylic acid (5). Method I. Ester 4 (1.0 g, 4.8 mmol) was added to a solution of NaOH (0.77 g, 19.0 mmol) in water (10 ml), the temperature was increased to 80°C and maintained for 10 h. The reaction mixture was cooled, acidified with HCl to pH 1, the precipitate that formed was filtered off, washed with H2O, and air-dried. The filtrate was extracted with Et2O (2×20 ml), dried over anhydrous Na2SO4, the solvent was removed and the solid residue was combined with the precipitate obtained above. Acid 5 was obtained as a white powder (0.83 g, 89% yield), mp 255–257°C (CHCl3–MeOH, 4:1). IR spectrum, ν, cm−1: 3221 (s), 3124 (m), 3008 (m), 2919 (m), 1699 (s), 1452 (w), 1292 (w), 1225 (m), 1203 (m), 1009 (w), 954 (m), 894 (w), 856 (w), 781 (w), 744 (w). 1H NMR spectrum, δ, ppm: 14.46 (2H, br. s, NН, ОН); 7.23 (1H, s, H-4); 2.60 (3H, s, CH3). 13C NMR spectrum, δ, ppm: 160.1 (С=О); 150.4; 148.2; 139.1; 136.7; 108.4 (C-4); 9.4 (CH3). Mass spectrum, m/z: 194 [M]+, 176 [M−H2O]+. Found, %: C 42.56; H 2.96; N 28.20. С7H6N4O3·0.2Н2О. Calculated, %: C 42.52; H 3.26; N 28.33.
Method II. A solution of a mixture of isomers 7a and 7b (0.91 g, 4.1 mmol) in anhydrous THF (15 ml) at −55°C was treated by the addition of a hexane solution of n-BuLi (2.5 ml, 6.2 mmol, 2.5 М). The reaction mixture was stirred for 30 min, followed by bubbling of CO2 at −50°C over the course of 10–15 min. Then the reaction mixture was warmed to room temperature, stirred for 2 h, and worked up by adding water (5 ml) and hexane (15 ml). The aqueous layer containing the product was separated, acidified with concd HCl (0.62 ml), extracted with EtOAc (3×15 ml), the combined extracts were dried over anhydrous Na2SO4, then the solvent was removed, leaving a solid residue of product 5. Yield 0.62 g (78%), colorless crystals, mp 255–257°C (Н2O). The sample was identical to the product obtained according to the method I.
3-[1-(1-Ethoxyethyl)-1 Н -pyrazol-3-yl]-4-methyl-1,2,5-oxadiazole (7a) and 3-[1-(1-ethoxyethyl)-1 Н -pyrazol-5-yl]-4-methyl-1,2,5-oxadiazole (7b). A solution of acid 5 (0.75 g, 5.0 mmol) in anhydrous CH2Cl2 (5 ml) was stirred and treated by the addition of CF3CO2H (60 mg, 0.53 mmol), followed by dropwise addition of vinyl ethyl ether (0.62 ml, 6.4 mmol), and the mixture was stirred at room temperature for 24 h. The reaction mixture was washed with saturated aqueous NaHCO3 solution (1.5 ml) and water (1.5 ml), the organic layer was dried over anhydrous Na2SO4, the solvent was removed and a 9:1 mixture of compounds 7a and 7b (0.91 g, 82% yield) was isolated as a thick oil. 1H NMR spectrum, δ, ppm (J, Hz): 8.16 (1Н, d, J = 2.4, Н 5, 7a); 7.82 (1Н, d, J = 2.0, Н-3, 7b); 6.94 (1Н, d, J = 2.0, Н-4, 7b); 6.91 (1Н, d, J = 2.4, Н-4, 7a); 6.00 (1Н, q, J = 5.9, СНСН3, 7b); 5.68 (1Н, q, J = 5.9, СНСН3,7a); 3.50–3.20 (2Н, m, СН 2СН3, 7a,b); 2.62 (3Н, s, СН3 furazan, 7a); 2.49 (3Н, s, СН3 furazan, 7b); 1.67 (3Н, d, J = 5.9, СНСН 3, 7a); 1.64 (3Н, d, J = 5.9, СНСН 3,7b); 1.06 (3Н, t, J = 6.8, СН2СН 3,7a); 0.95 (3Н, t, J = 6.8, СН2СН 3, 7b).
[4-(1 H -Pyrazol-3(5)-yl)-1,2,5-oxadiazol-3-yl]acetic acid (8). A solution of pyrazole 6 (5.0 g, 33.0 mmol) in anhydrous THF (100 ml) was stirred at −55°C and treated by the addition of hexane solution of n-BuLi (26.5 ml, 66.0 mmol, 2.5 М), followed by stirring of the reaction mixture for 10–15 min. Then CO2 was bubbled through the reaction mixture for 10–15 min at −50°C, the reaction mixture was warmed to room temperature while stirring for additional 2 h and worked up by adding water (30 ml) and hexane (100 ml). The aqueous layer containing the product was separated, acidified with concd HCl (6.7 ml), and extracted with EtOAc (3×100 ml). The combined extracts were dried over anhydrous Na2SO4, then the solvent was removed by evaporation at reduced pressure, giving a solid milky-white residue. Yield 2.6 g (41%), colorless crystals, mp 161–163°C (EtOAc–CCl4, 1:1). IR spectrum, ν, cm−1: 3312 (s), 3160 (m), 3141 (m), 3038 (w), 2933 (w), 2756 (w), 2652 (w), 2617 (w), 2560 (w), 1728 (vs), 1525 (w), 1439 (m), 1396 (w), 1384 (w), 1331 (m), 1290 (m), 1244 (s), 1181 (w), 1129 (w), 1075 (w), 1052 (w), 957 (m), 923 (m), 895 (m), 787 (s), 729 (m), 662 (w), 608 (m), 412 (m). 1H NMR spectrum, δ, ppm (J, Hz): 13.40 (1H, br. s, NН); 12.91 (1H, br. s, ОН); 7.99 (1H, d, J = 2.1, H-3); 6.87 (1H, d, J = 2.1, H-4); 4.22 (2H, s, CH2). 13C NMR spectrum, δ, ppm: 169.6 (С=О); 149.2 (С furazan); 148.9 (С furazan); 138.1 (С-3); 136.7 (C-5); 108.4 (C-4); 30.1 (CH2). Mass spectrum, m/z: 194 [M]+. Found, m/z: 195.0514 [M+H]+. С7H7N4O3. Calculated, m/z: 195.0513. Found, %: C 43.06; H 3.10; N 28.47. С7H6N4O3. Calculated, %: C 43.30; H 3.11; N 28.86.
3-Methyl-4-(4-nitro-1 H -pyrazol-5-yl)-1,2,5-oxadiazole (9). A suspension of acid 5 (0.25 g, 1.3 mmol) in H2SO4 (2.6 ml, ρ 1.82 g/cm3) was treated by dropwise addition of HNO3 (0.17 ml, ρ 1.50 g/cm3) and the mixture was stirred for 2 h at 80–85°C. Then the reaction mixture was cooled, poured into ice water (10 ml); the precipitate that formed was filtered off, washed with water, and air-dried. Yield 0.13 g (52%), white powder, mp 161–162°C (CНCl3–МеОН, 4:1) (mp 161–162°C15). 1H NMR spectrum, δ, ppm: 14.64 (1H, br. s, NН); 9.14 (1H, s, H-5); 2.38 (3H, s, CH3).
Methyl 3(5)-(4-methyl-1,2,5-oxadiazol-3-yl)-1-nitro-1 Н -pyrazole-5(3)-carboxylates 10a,b. A solution of ester 3 (0.53 g, 2.6 mmol) in glacial acetic acid (2.5 ml) was cooled to 5–10°C and treated by dropwise addition of HNO3 (0.5 ml, ρ 1.50 g/cm3), followed by the addition of acetic anhydride (1.3 ml). The mixture was stirred at the same temperature for 6 h, then poured into ice water (25 ml); the precipitate that formed was filtered off and washed with water. The filtrate was extracted with CH2Cl2 (3×15 ml), the organic layer was dried over anhydrous CaCl2, the solvent was removed. A 1:1 mixture of isomers 10a and 10b (0.58 g, 90%) was isolated as a white powder. IR spectrum, ν, cm−1: 3228 (w), 1732 (s), 1644 (s), 1444 (w), 1384 (w), 1284 (s), 1256 (s), 1148 (s), 1016 (w), 944 (w), 896 (w), 844 (w), 824 (w), 796 (w), 780 (w). 1H NMR spectrum (acetone-d 6), δ, ppm: 7.35 (1Н, s, H-4); 3.95 (3Н, s, ОCH3); 2.67 (3Н, s, CH3). 1H NMR spectrum (CDCl3), δ, ppm: 7.39 (1Н, s, H-4, 10b); 7.25 (1H, s, H-4, 10a); 4.02 (3Н, s, ОCH3); 2.67 (3Н, s, CH3). 13C NMR spectrum (CDCl3), δ, ppm: 160.1 (C=O, 10b); 157.6 (C=O, 10a); 150.9 (С furazan, 10b); 150.2 (С furazan, 10a); 146.3 (С furazan, 10a); 144.8 (С furazan, 10b); 141.7 (10b); 138.8 (10a); 133.0 (10a); 126.8 (10b); 115.1 (C-4, 10b); 112.7 (C-4, 10a); 53.9 (OCH3, 10a); 53.3 (OCH3, 10b); 9.6 (CH3, 10a); 8.5 (CH3, 10b). 14N NMR spectrum (CDCl3), δ, ppm: −63.53 (N–NO2). Found, m/z: 276.0350 [M+Na]+. С8H9N5NaO5. Calculated, m/z: 276.0339.
Methyl 5-(4-methyl-1,2,5-oxadiazol-3-yl)-4-nitro-1 Н -pyrazole-3-carboxylate (11). A solution of nitro acid 1 (3.92 g, 16.4 mmol) in MeOH (25 ml) was treated by dropwise addition of SOCl2 (1.4 ml, 19.3 mmol) and the mixture was stirred at 60–65°C for 3 h. The reaction mixture was cooled, the solvent was removed by evaporation at reduced pressure and the residue was poured into H2O. The obtained white precipitate was then filtered, washed with H2O, and air-dried. Yield 3.73 g (90%), white powder, mp 109–111°C (H2O–EtOH, 1:1). IR spectrum, ν, cm−1: 3168 (m), 1739 (s), 1535 (s), 1449 (w), 1375 (m), 1311 (m), 1250 (s), 1221 (m), 1091 (w), 980 (s), 907 (w), 835 (m), 783 (w). 1H NMR spectrum, δ, ppm: 3.95 (3Н, s, OCH3); 2.53 (3Н, s, CH3). 13C NMR spectrum, δ, ppm: 157.3 (С=О); 151.4 (С furazan); 145.7 (С furazan); 133.1; 131.4; 131.3; 53.4 (OCH3); 8.8 (CH3). Mass spectrum, m/z: 253 [M]+. Found, %: C 37.91; H 2.68; N 27.53. С8H7N5O5. Calculated, %: C 37.95; H 2.79; N 27.66.
5-(4-Methyl-1,2,5-oxadiazol-3-yl)-4-nitro-1 Н -pyrazole-3-carboxamide (12). A solution of nitro ester 11 (4.92 g, 19.4 mmol) in MeOH (15 ml) was treated by the addition of aqueous 19% ammonia solution (20 ml, 0.2 mol, ρ 0.926 g/cm3). The mixture was maintained for 24 h at 40°C. The solvent was removed by evaporation at reduced pressure, the residue was suspended in water, acidified with HCl to pH 1, the precipitate was filtered off, washed with water, and air-dried. The filtrate was extracted with EtOAc (3×30 ml), the extract was dried over anhydrous Na2SO4, the solvent was removed by evaporation at reduced pressure, giving a solid residue. Both portions of the solid product were combined. Yield 4.25 g (92%), mp 230–232°C (CHCl3–MeOH, 4:1). IR spectrum, ν, cm−1: 3376 (m), 3231 (m), 1687 (s), 1522 (s), 1449 (m), 1375 (w), 1337 (m), 1218 (w), 1145 (w), 973 (w), 838 (w). 1H NMR spectrum, δ, ppm: 15.20 (1Н, br. s, NН); 8.39 (1Н, s, NH); 8.26 (1Н, s, NH); 2.39 (3Н, s, CH3). 13C NMR spectrum, δ, ppm: 158.4 (С=О); 151.9 (С furazan); 146.8 (С furazan); 138.9 (C-3); 131.9 (br. s, С-4); 130.6 (C-5); 8.2 (CH3). Mass spectrum, m/z: 237 [M−H]+, 222 [M−NH2]+. Found, %: C 30.84; H 3.85; N 30.70. С7H6N6O4×2H2O. Calculated, %: C 30.66; H 3.68; N 30.65.
5-(4-Methyl-1,2,5-oxadiazol-3-yl)-4-nitro-1 Н -pyrazol-3-amine (13). Elemental bromine (0.13 ml, 2.5 mmol) was added dropwise to a solution of NaOH (0.48 g, 12.0 mmol) in water (10 ml) at 0–5°C. Amide 12 (0.5 g, 2.1 mmol) was added portionwise to the obtained solution, and the reaction mixture was stirred for additional 40 min at 0–5°C, then the temperature was increased to 55–60°C and maintained for 2 h. After cooling to 10°C, the mixture was acidified with HCl to pH 2–3. The precipitate that formed was filtered off, washed with water, and air-dried. The filtrate was extracted with CH2Cl2 (2×50 ml) and dried over anhydrous CaCl2. The solvent was removed by evaporation at reduced pressure, giving a solid residue that was combined with the sample obtained above. Yield 0.42 g (95%), light-yellow powder, mp 254–256°C (CHCl3–MeOH, 3:1). IR spectrum, ν, cm−1: 3425 (s), 3313 (m), 3218 (s), 1652 (s), 1457 (m), 1444 (s), 1404 (m), 1365 (s), 1334 (s), 1204 (m), 988 (m), 975 (m), 890 (w), 831 (w), 757 (m), 744 (w). 1H NMR spectrum, δ, ppm: 13.04 (1Н, br. s, NН); 7.59 (2Н, s, NH2); 2.35 (3Н, s, CH3). 13C NMR spectrum, δ, ppm: 152.5 (С furazan); 148.8 (2С); 132.7 (С-5); 116.8 (br. s, С-4); 8.6 (CH3). 14N NMR spectrum, δ, ppm: −20.25. Found, m/z: 211.0573 [M+H]+. С6H7N6O3. Calculated, m/z: 211.0574.
5-(4-Methyl-1,2,5-oxadiazol-3-yl)- N ,4-dinitro-1 Н -pyrazol-3-amine (14). A solution of nitroaminopyrazole 13 (1.05 g, 5.0 mmol) in CF3CO2H (5 ml) was cooled to 5–10°C and treated by dropwise addition of HNO3 (0.5 ml, 50.0 mmol, ρ 1.50 g/cm3), followed by acetic anhydride (0.94 ml, 10.0 mmol). The reaction mixture was stirred at the same temperature for 10 h. The mixture was then poured into ice water (30 ml), the precipitate that formed was filtered off and washed with H2O. The filtrate was extracted with Et2O (4×25 ml), the organic layer was dried over anhydrous Na2SO4, the solvent was removed by evaporation at reduced pressure, giving a solid residue. Both portions of the solid product were combined and crystallized from water. Yield 0.98 g (77%), yellow precipitate, mp 90–92°C. IR spectrum, ν, cm−1: 3456 (m), 3396 (m), 3308 (m), 1616 (s), 1516 (s), 1492 (s), 1376 (m), 1352 (s), 1332 (s), 1296 (s), 1216 (s), 1016 (w), 976 (w), 908 (w), 872 (w), 636 (w). 1H NMR spectrum (acetone-d 6), δ, ppm: 2.47 (3Н, s, CH3). 13C NMR spectrum (acetone-d 6), δ, ppm: 152.7 (С furazan); 146.3 (С furazan); 138.1 (C-3, NHNO2); 132.1 (C-5); 131.6 (br. s, C-4); 8.3 (CH3). 14N NMR spectrum (acetone-d 6), δ, ppm: −23.33 (C–NO2); −35.78 (NHNO2). Found, m/z: 254.0274 [M−H]−. С6H4N7O5. Calculated, m/z: 254.0279.
References
(а) Zaitsev, A. A.; Dalinger, I. L.; Shevelev, S. A. Russ. Chem. Rev. 2009, 78, 589. [Usp. Khim. 2009, 643.] (b) Shevelev, S. A.; Dalinger, I. L. Russ. J. Org. Chem. 1998, 34, 1071. [Zh. Org. Khim. 1998, 34, 1127.]
Bakavoli, M.; Bagherzadeh, G.; Vaseghifar, M.; Shiri, A.; Pordel, M.; Mashreghi, M.; Pordeli, P.; Araghi, M. Eur. J. Med. Chem. 2010, 45, 647.
(а) Rawson, D. J.; Ballard, S.; Barber, C.; Barker, L.; Beaumont, K.; Bunnage, M.; Cole, S.; Corless, M.; Denton, S.; Ellis, D.; Floc'h, M.; Foster, L.; Gosset, J.; Holmwood, F.; Lane, C.; Leahy, D.; Mathias, J.; Maw, G.; Million, W.; Poinsard, C.; Price, J.; Russel, R.; Street, S.; Watson, L. Bioorg. Med. Chem. 2012, 20, 498. (b) DeNinno, M. P.; Andrews, M.; Bell, A. S.; Chen, Y.; Eller-Zarbo, C.; Eshelby, N.; Etienne, J. B.; Moore, D. E.; Palmer, M. J.; Visser, M. S.; Yu, L. J.; Zavadovski, W. J.; Gibbs, E. M. Bioorg. Med. Chem. Lett. 2009, 19, 2537. (c) Allerton, C. M. N.; Barber, C. G.; Beaumont, K. C.; Brown, D. G.; Cole, S. M.; Ellis, D.; Lane, C. A. L.; Maw, G. N.; Mount, N. M.; Rawson, D. J.; Robinson, C. M.; Street, S. D. A.; Summerhill, N. W. J. Med. Chem. 2006, 49, 3581.
(а) Sheikhi-Mohammareh, S.; Shiri, A.; Bakavoli, M.; Mague, J. J. Heterocycl. Chem. 2016, 53, 1231. (b) Dalinger, I. L.; Vatsadse, I. A.; Shevelev, S. A.; Ivachtchenko, A. V. J. Comb. Chem. 2005, 7, 236.
Baraldi, P. G.; Garuti, L.; Roberti, M. Synthesis 1994, 1437.
(а) Sun, S.; Zhang, Z.; Pokrovskaia, N.; Chowdhury, S.; Jia, Q.; Chang, E.; Khakh, K.; Kwan, R.; McLaren, D. G.; Radomski, C. C.; Ratkay, L. G.; Fu, J.; Dales, N. A.; Winther, M. D. Bioorg. Med. Chem. 2015, 23, 455. (b) Yasuda, Y.; Arakawa, T.; Nawata, Y.; Shimada, S.; Oishi, S.; Fujii, N.; Nishimura, S.; Hattori, A.; Kakeya, H. Bioorg. Med. Chem. 2015, 23, 1776. (c) Ward, R. A.; Anderton, M. J.; Ashton, S.; Bethel, P. A.; Box, M.; Butterworth, S.; Colclough, N.; Chorley, C. G.; Chuaqui, C.; Cross, D. A. E.; Dakin, L. A.; Debreczeni, J. É.; Eberlein, C.; Finlay, M. R. V.; Hill, G. B.; Grist, M.; Klinowska, T. C. M.; Lane, C.; Martin, S.; Orme, J. P.; Smith, P.; Wang, F.; Waring, M. J. J. Med. Chem. 2013, 56, 7025. (d) Plummer, M. S.; Cornicelli, J.; Roark, H.; Skalitzky, D. J.; Stankovic, C. J.; Bove, S.; Pandit, J.; Goodman, A.; Hicks, J.; Shahripour, A.; Beidler, D.; Lu, X. K.; Sanchez, B.; Whitehead, C.; Sarver, R.; Braden, T.; Gowan, R.; Shen, X. Q.; Welch, K.; Ogden, A.; Sadagopan, N.; Baum, H.; Miller, H.; Banotai, C.; Spessard, C.; Lightle, S. Bioorg. Med. Chem. Lett. 2013, 23, 3438.
(а) Machado, P.; Lima, G. R.; Rotta, M.; Bonacorso, H. G.; Zanatta, N.; Martins, M. A. P. Ultrason. Sonochem. 2011, 18, 293. (b) Skinner, P. J.; Webb, P. J.; Sage, C. R.; Dang, H. T.; Pride, C. C.; Chen, R.; Tamura, S. Y.; Richman, J. G.; Connolly, D. T.; Semple, G. Bioorg. Med. Chem. Lett. 2009, 19, 4207. (c) Zhang, Y.; Burgess, J. P.; Brackeen, M.; Gilliam, A.; Mascarella, S. W.; Page, K.; Seltzman, H. H.; Thomas, B. F. J. Med. Chem. 2008, 51, 3526. (d) Lee, S. H.; Seo, H. J.; Lee, S.-H.; Jung, M. E.; Park, J.-H.; Park, H.-J.; Yoo, J.; Yun, H.; Na, J.; Kang, S. Y.; Song, K.-S.; Kim, M.-a.; Chang, C.-H.; Kim, J.; Lee, J. J. Med. Chem. 2008, 51, 7216. (e) Varano, F.; Catarzi, D.; Colotta, V.; Filacchioni, G.; Galli, A.; Costagli, C.; Carlà, V. J. Med. Chem. 2002, 45, 1035.
(а) Hochdörffer, K.; März-Berberich, J.; Nagel-Steger, L.; Epple, M.; Meyer-Zaika, W.; Horn, A. H. C.; Sticht, H.; Sinha, S.; Bitan, G.; Schrader, T. J. Am. Chem. Soc. 2011, 133, 4348. (b) Rzepecki, P.; Hochdörffer, K.; Schaller, T.; Zienau, J.; Harms, K.; Ochsenfeld, C.; Xie, X.; Schrader, T. J. Am. Chem. Soc. 2008, 130, 586. (c) Kusakiewicz-Dawid, A.; Górecki, Ł.; Masiukiewicz, E.; Rzeszotarska, B. Synth. Commun. 2009, 39, 4122. (d) Rzepecki, P.; Wehner, M.; Molt, O.; Zadmard, R.; Harms, K.; Schrader, T. Synthesis 2003, 1815.
Huang, Z.; Tong, J.; Zhou, S.; Xiong, L.; Wang, H.; Zhao, Y. J. Heterocycl. Chem. 2016, 53, 1036.
Zhang, D. Q.; Xu, G. F.; Fan, Z. J.; Wang, D. Q.; Yang, X. L.; Yuan, D. K. Chin. Chem. Lett. 2012, 23, 669.
(а) Dalinger, I. L.; Vatsadse, I. A.; Shkineva, T. K.; Popova, G. P.; Ugrak, B. I.; Shevelev, S. A. Russ. Chem. Bull., Int. Ed. 2010, 59, 1631. [Izv. Akad. Nauk, Ser. Khim. 2010, 1589.] (b) Shevelev, S. A.; Vinogradov, V. M.; Dalinger, I. L.; Cherkasova, T. I. Russ. Chem. Bull. 1993, 42, 1861. [Izv. Akad. Nauk, Ser. Khim. 1993, 1945.]
Ek, S.; Latypov, N. J. Heterocycl. Chem. 2014, 51, 1621.
Latypov, N. V.; Silevich, V. A.; Ivanov, P. A.; Pevzner, M. S. Chem. Heterocycl. Compd. 1976, 12, 1355. [Khim. Geterotsikl. Soedin. 1976, 1649.]
(а) Pagoria, P. Propellants, Explos., Pyrotech. 2016, 41, 452. (b) Klapötke, T. M.; Witkowski, T. G. Propellants, Explos., Pyrotech. 2016, 41, 470.
Dalinger, I. L.; Vatsadze, I. A.; Shkineva, T. K.; Kormanov, A. V.; Kozeev, A. M.; Averkiev, B. B.; Dalinger, A. I.; Beklemishev, M. K.; Sheremetev, A. B. Chem. Heterocycl. Compd. 2015, 51, 545. [Khim. Geterotsikl. Soedin. 2015, 51, 545.]
(а) Dalinger, I. L.; Suponitsky, K. Yu.; Pivkina, A. N.; Sheremetev, A. B. Propellants, Explos., Pyrotech. 2016, 41, 789. (b) Dalinger, I. L.; Kormanov, A. V.; Vatsadze, I. A.; Shkineva, T. K.; Kozeev, A. M.; Averkiev, B. B.; Sheremetev, A. B. Chem. Heterocycl. Compd. 2015, 51, 819. [Khim. Geterotsikl. Soedin. 2015, 51, 819.]
(а) Vatsadze, I. A.; Serushkina, O. V.; Dutov, M. D.; Shkineva, T. K.; Suponitsky, K. Yu.; Ugrak, B. I.; Dalinger, I. L. Chem. Heterocycl. Compd. 2015, 51, 695. [Khim. Geterotsikl. Soedin. 2015, 51, 695.] (b) Palysaeva, N. V.; Kumpan, K. P.; Struchkova, M. I.; Dalinger, I. L.; Kormanov, A. V.; Aleksandrova, N. S.; Chernyshev, V. M.; Pyreu, D. F.; Suponitsky, K. Yu.; Sheremetev, A. B. Org. Lett. 2014, 16, 406.
Dalinger, I. L.; Kormanov, A. V.; Vatsadze, I. A.; Serushkina, O. V.; Shkineva, T. K.; Suponitsky, K. Yu.; Pivkina, A. N.; Sheremetev, A. B. Chem. Heterocycl. Compd. 2016, 52, 1025. [Khim. Geterotsikl. Soedin. 2016, 52, 1025.]
(а) Perevalov, V. P.; Manaev, Yu. A.; Baryshnenkova, L. I.; Kanep, E. É.; Andreeva, M. A.; Stepanov, B. I. Chem. Heterocycl. Compd. 1987, 23, 1081. [Khim. Geterotsikl. Soedin. 1987, 1350.] (b) Manaev, Yu. A.; Andreeva, M. A.; Perevalov, V. P.; Stepanov, B. I.; Dubrovskaya, V. A.; Seraya, V. I. Zh. Obsch. Khim. 1982, 2592.
Zaitsev, A. A.; Cherkasova, T. I.; Dalinger, I. L.; Kachala, V. V.; Strelenko, Yu. A.; Fedyanin, I. V.; Solkan, V. N.; Shkineva, T. K.; Popova, G. P.; Shevelev, S. A. Russ. Chem. Bull., Int. Ed. 2007, 56, 2074. [Izv. Akad. Nauk, Ser. Khim. 2007, 2004.]
Shevelev, S. A.; Dalinger, I. L.; Shkineva, T. K.; Ugrak, B. I.; Gulevskaya, V. I.; Kanishchev, M. I. Russ. Chem. Bull. 1993, 42, 1063. [Izv. Akad. Nauk, Ser. Khim. 1993, 1108.]
Zhang, J.; Parrish, D. A.; Shreeve, J. M. Chem.–Asian J. 2014, 9, 2953.
Li, Y.; Shu, Y.; Wang, B.; Zhang, S.; Zhai, L. RSC Adv. 2016, 6, 84760.
(а) Khidre, R. E.; Abdel-Wahab, B. F.; Farahat, A. A.; Mohamed, H. A. J. Heterocycl. Chem. 2016, 53, 13. (b) Nolsöe, J. M. J.; Weigelt, D. J. Heterocycl. Chem. 2009, 46, 1.
Kulikov, A. S.; Makhova, N. N.; Godovikova, T. I.; Golova, S. P.; Khmel'nitskii, L. I. Russ. Chem. Bull. 1994, 43, 630. [Izv. Akad. Nauk, Ser. Khim. 1994, 679.]
(а) Bagal, S. K.; Bungay, P. J.; Denton, S. M.; Gibson, K. R.; Glossop, M. S.; Hay, T. L.; Kemp, M. I.; Lane, C. A. L.; Lewis, M. L.; Maw, G. N.; Million, W. A.; Payne, C. E.; Poinsard, C.; Rawson, D. J.; Stammen, B. L.; Stevens, E. B.; Thompson, L. R. ACS Med. Chem. Lett. 2015, 6, 650. (b) Schlosser, M.; Volle, J.-N.; Leroux, F.; Schenk, K. Eur. J. Org. Chem. 2002, 2913.
Roy, S.; Roy, S.; Gribble, G. W. Top. Heterocycl. Chem. 2012, 29, 155.
Catalan, J.; Elguero, J. Adv. Heterocycl. Chem. 1987, 41, 187.
(а) Sizov, A. Yu.; Sheremetev, A. B. Russ. Chem. Bull. 1992, 41, 281. [Izv. Akad. Nauk, Ser. Khim. 1992, 365.] (b) Sheremetev, A. B.; Ivanova, E. A.; Sizov, A. Yu.; Kulagina, V. O.; Dmitriev, D. E.; Strelenko, Yu. A. Russ. Chem. Bull., Int. Ed. 2003, 52, 679. [Izv. Akad. Nauk, Ser. Khim. 2003, 651.] (c) Sheremetev, A. B.; Ivanova, E. A. Russ. Chem. Bull., Int. Ed. 2003, 52, 2017. [Izv. Akad. Nauk, Ser. Khim. 2003, 1910.] (d) Sheremetev, A. B.; Ivanova, E. A.; Shatunova, E. V.; Dmitriev, D. E.; Kuz'mina, N. E. Russ. Chem. Bull., Int. Ed. 2004, 53, 615. [Izv. Akad. Nauk, Ser. Khim. 2004, 587.] (e) Sheremetev, A. B.; Ivanova, E. A.; Spiridonova, N. P.; Melnikova, S. F.; Tselinsky, I. V.; Suponitsky, K. Yu.; Antipin, M. Yu. J. Heterocycl. Chem. 2005, 42, 1237.
(а) Boyer, J. H. Nitroazoles: The C-Nitro Derivatives of Five-Membered N- and N,O-Heterocycles (Organic Nitro Chemistry Series); VCH: Weinheim, 1986. (b) Larina, L.; Lopyrev, V. Nitroazoles. Synthesis, Structure and Applications; Springer: LLC, New York, 2009.
Perevalov, V. P.; Manaev, Yu. A.; Andreeva, M. A.; Stepanov, B. I. Zh. Obsch. Khim. 1985, 55, 882.
Vinogradov, V. M.; Cherkasova, T. I.; Dalinger, I. L.; Shevelev, S. A. Russ. Chem. Bull. 1993, 42, 1552. [Izv. Akad. Nauk, Ser. Khim. 1993, 1616.]
(а). Grigor'ev, N. B.; Levina, V. I.; Shevelev, S. A.; Dalinger, I. L.; Granik, V. G. Mendeleev Commun. 1996, 6, 11. (b). Grigor'ev, N. B.; Kalinkina, M. I.; Chechekin, G. V.; Nikitin, V. B.; Engalycheva, G. N.; Belushkina, N. N.; Levina, V. I.; Dalinger, I. L.; Mashkovskii, M. D.; Shevelev, S. A.; Litosh, V. A.; Vener, M. V.; Severina, I. S.; Kaminka, M. É.; Arzamastsev, A. P.; Granik, V. G. Pharm. Chem. J. 1998, 32, 127. [Khim.-Farm. Zh. 1998, 32(3), 15.]
Xuan, B.; Wang, T.; Chiou, G. C. Y; Dalinger, I.; Shkineva, T. K.; Shevelev, S. A. J. Ocul. Pharmacol. Ther. 2001, 17, 505.
Habraken, C. L.; Janssen, J. W. A. M. J. Org. Chem. 1971, 36, 3081.
Yin, P.; Parrish, D. A.; Shreeve, J. M. J. Am. Chem. Soc. 2015, 137, 4778.
Sheremetev, A. B. Ross. Khim. Zh. 1997, 43. [Mendeleev Chem. J. 1997, 41, 62.]
(а) Yin, P.; Zhang, Q.; Shreeve, J. M. Acс. Сhem. Res. 2016, 49, 4. (b) Liu, Y.; Zhang, J.; Wang, K.; Li, J.; Zhang, Q.; Shreeve, J. M. Angew. Chem., Int. Ed. 2016, 55, 11548. (c) Fischer, D.; Gottfried, J. L.; Klapötke, T. M.; Karaghiosoff, K.; Stierstorfer, J.; Witkowski, T. G. Angew. Chem., Int. Ed. 2016, 55, 16132. (d) He, C.; Tang, Y.; Mitchell, L. A.; Parrish, D. A.; Shreeve, J. M. J. Mater. Chem. A 2016, 4, 8969. (e) Klapötke, T. M.; Leroux, M.; Schmid, P. C.; Stierstorfer, J. Chem.–Asian J. 2016, 11, 844. (f) Qu, Y.; Zeng, Q.; Wang, J.; Ma, Q.; Li, H.; Li, H.; Yang, G. Chem.–Eur. J. 2016, 22, 12527.
The work was performed with financial support from the Russian Science Foundation (project RNF 14-13-01153).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2017, 53(8), 876–882
Rights and permissions
About this article

Cite this article
Kormanov, A.V., Lipilin, D.L., Shkineva, T.K. et al. Synthesis and transformations of 3(5)-(3-methylfurazan-4-yl)-4-nitro-1Н-pyrazole-5(3)-carboxylic acid. Chem Heterocycl Comp 53, 876–882 (2017). https://doi.org/10.1007/s10593-017-2141-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-017-2141-6