

A new series of 3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl]-4H-chromen-4-ones has been synthesized from substituted 2-hydroxyacetophenones and 1-phenyl-3-(thiophen-2-yl)-1H-pyrazole-4-carbaldehyde using NaOH and H2O2 by modified Algar–Flynn–Oyamada reaction under conventional and microwave irradiation conditions. In this method flavonols are synthesized without isolating chalcones in good yields (80–85%). The structures of the compounds were established on the basis of IR, 1H, 13C NMR and mass spectral and elemental analysis data. The synthesized compounds were screened for their antibacterial and antifungal activities.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
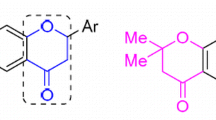
Heterocyclic compounds containing nitrogen and oxygen play important role in the design of agrochemicals and pharmaceuticals. Pyrazole, a five-membered aromatic nitrogen heterocycle, forms a part of the structure of several natural products and drugs.1 – 3 Pyrazole derivatives are known to possess antimicrobial,4 antiviral,3 antitumor,5 , 6 antihypertensive,7 antidepressant,8 insecticidal,9 antifungal,10 5α-reductase inhibitory,11 antiproliferative,12 antiparasitic,13 herbicidal,14 anti-inflammatory,15 , 16 antiprotozoal,17 analgesic,16 and androgen receptor modulatory18 activities. The pyrazole ring is present as the core in a variety of leading drugs such as lonazolac19 and rimonabant20 (Fig. 1).

Structures of some biologically active pyrazoles, thiophenes, and flavonols.
Thiophene structure can be found in certain natural products and is also incorporated in several pharmacologically active compounds. In medicinal chemistry, thiophene derivatives are very well known for their therapeutic applications, e.g., tienilic acid, a loop diuretic drug,21 and [5-ethyl-6-methyl-2-(methylsulfanyl)thieno[2,3-d]pyrimidin-4-yl]hydrazine, an antimicrobial agent22 (Fig. 1).
Flavanoids are a group of common and naturally occurring compounds that are widely found in the plant kingdom.23 They occur naturally as plant pigments in many fruits and vegetables, as well as beverages, such as tea, red wine, coffee, and beer.24 Flavonols are a class of flavonoids that have the 3-hydroxychromone backbone. Synthesis of flavonol derivatives is of great interest and has a long history.25 These compounds form a major class of naturally occuring compounds and interest in their chemistry is on a higher scale because of the broad spectrum of biological activities associated, such as free radical scavenging,26 antimicrobial,27 cytotoxic,28 neuroprotective,29 and antioxidant.30 The most studied flavonols are quercetin, myricetin, and kaempferol (Fig. 1) which found to exhibit various biological activities including anticancer activity.31
Inspired by the pharmacological profile of all these components, in this work we planned to synthesize 2-hetaryl-substituted 3-hydroxychromones including in their structure pyrazole and thiophene cycles. For the formation of 3-hydroxy-4H-chromen-4-one derivatives, the modified Algar–Flynn–Oyamada (AFO) reaction has got considerable attention. The original AFO reaction32 is a twostep process for the synthesis of 3-hydroxychromenones. In the first step, formation of 2-hydroxychalcones takes place which on subsequent cyclization in second step in the presence of alkaline hydrogen peroxide yields the corresponding flavonols. Whereas the modified AFO reaction33 is a one-step process for the synthesis of flavonols from 2-hydroxyacetophenone and aromatic aldehydes in the presence of alkaline hydrogen peroxide. In this method there is no need to isolate the intermediate chalcones, which reduces the time, cost, and amounts of chemicals used. Microwave irradiation was also employed for heating the reaction mixture because it provides instantaneous and very specific heating without contact between the energy source and the reaction vessel.34
Based on these facts, we have taken up microwave-assisted synthesis of substituted 3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl]-4H-chromen-4-ones 3a–h by modified AFO reaction. The obtained products were tested for their antimicrobial activity.
As a model reaction, 2-hydroxyacetophenone (1a) was condensed with 1-phenyl-3-(thiophen-2-yl)-1H-pyrazole-4-carbaldehyde (2) using alkali and subsequently oxidized with alkaline hydrogen peroxide to yield flavonol derivative.
The model reaction was carried out in conventional as well as in microwave irradiation conditions (Scheme 1, Table 1). The product was identified by IR, 1H, 13C NMR and mass spectral data as 3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl]-4H-chromen-4-one (3a), that ruled out the formation of corresponding aurones and some other benzofuran derivatives which were supposed to form as by-products in AFO reaction. This assumption of the reaction mechanism and cyclization of the chalcone were supported by literature.35 In the IR spectrum of compound 3a, the characteristic flavonol carbonyl group absorption observed at 1606 cm–1 and hydroxy group absorption observed at 3161 cm–1. In the 1H NMR spectrum of compound 3a, the OH proton appeared as a singlet at 7.28 ppm. The pyrazole proton appeared as a singlet at 8.79 ppm. The proton H-5 was appeared as a doublet (J = 6.8 Hz) at 8.25 ppm. In 13C NMR spectrum (75 MHz, CDCl3) of compound 3a, the carbonyl carbon appeared at 172.5 ppm. These values are in close agreement with the previously reported values for the respective flavonol carbons.36 In the mass spectrum (ESI) of compound 3a the base peak was observed at m/z 387 corresponding to [M+H]+ ion.

Scheme 1
The remaining compounds 3b–h were synthesized in the similar way using both conventional and microwave conditions. We found that microwave irradiation provides much more faster conversion of the starting compounds and higher yields (reaction time 12–15 min, yields 80–87%) than conventional heating (reaction time 6–8 h, yields 50–58%).
All the compounds were screened for their antibacterial activity against Staphylococcus aureus, Bacillus subtilis, Pseudomonas aeruginosa, and Escherichia coli using ampicillin as the standard drug. The activity was determined using filter paper disc method by measuring the zone of inhibition in mm. The compounds were screened at the concentration of 50 μg/ml in DMSO. From the screening studies (Table 2), it is evident that the synthesized compounds 3e,g,h showed good antibacterial activity against all the tested organisms. Furthermore, changing the halogens from F to Cl or Br, did not provide any significant change in the levels of activity against bacteria. However, the activity does not depend much on the electronic nature of the compounds.
All the compounds were also screened for their antifungal activity against Aspergillus niger, Penicillium italicum, and Fusarium oxysporum using griseofulvin as the standard drug. The activity was determined using the cup plate agar diffusion method by measuring the zone of inhibition in mm. The compounds were screened at a concentration of 50 μg/ml in DMSO. From the screening studies (Table 3), it was evident that the synthesized compounds 3a,g,h showed good antifungal activity against all the tested organisms. The unsubstituted compound 3a showed the highest activity against the fungi. The highly electron-rich flavonols 3g,h showed activity which was comparable to that of compound 3a. Among the halo derivates, the bromosubstituted compound 3d showed significantly higher activity compared to those of the fluoro- and chlorosubstituted compounds 3b,c, respectively.
A series of some new 2-[1-phenyl-3-(thiophen-2-yl)-1Hpyrazol-4-yl]chromen-4-one derivatives have been synthesized by modified Algar–Flynn–Oyamada reaction under conventional and microwave irradiation conditions. All final compounds were investigated for their in vitro antimicrobial activity. Some of the obtained compounds show antimicrobial activity against selected microorganisms compared with the reference drugs. Thus, the above compounds can be considered as lead compounds for further development of more potent antimicrobial agents.
Experimental
IR spectra were recorded in KBr on a Shimadzu FTIR 8400S spectrophotometer. 1H and 13C NMR spectra were recorded on a Bruker Avance II 300 spectrometer (300 and 75 MHz, respectively) in CDCl3 using TMS as internal standard. Mass spectra were recorded on a Shimadzu LCMS-2020 mass spectrometer, electrospray ionization. Elemental analysis was carried out on a Vario-11 CHN analyzer. Melting points were determined in open glass capillaries on a Stuart SMP30 apparatus and are uncorrected. Purity of the compounds was checked by TLC on silica gel 60 F254 (Merck). All the microwave irradiation experiments were performed in a multiSYNTH series microwave system (Milestone). All solvents and chemicals were obtained commercially, mostly from Sigma-Aldrich, and were used without further purification.
Synthesis of substituted 3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1 H -pyrazol-4-yl]-4 H -chromen-4-ones 3a–f (General method).
I (Conventional heating). To a well-stirred solution of 2-hydroxyacetophenones 1a–h (1 mmol) and 1-phenyl-3-(thiophen-2-yl)-1H-pyrazole-4-carbaldehyde37 (2) (254 mg, 1 mmol) in EtOH (20 ml), a solution of NaOH (160 mg, 4 mmol) in EtOH (10 ml) was added at room temperature. The reaction mixture was further stirred for 4–5 h. After consumption of reactants (as indicated by TLC), the reaction mixture was dissolved in aqueous NaOH solution (200 mg, 5 mmol in 5 ml of H2O) and 30% H2O2 (3 ml) was added dropwise. Stirring was continued for 2–3 h. After completion of reaction (monitored by TLC), the resulting light-yellow reaction mixture was poured onto crushed ice and neutralized with dilute HCl. The light-yellow solid thus obtained was filtered off, washed with water, and dried. The crude product was purified by column chromatography on silica gel using hexane–EtOAc, 7:3, as eluent and further recrystallized from EtOH to afford the desired products 3a–h as yellow solids, the respective yields are shown in Table 1.
II (Microwave irradiation). To a well-stirred solution of 2-hydroxyacetophenones 1a–h (1 mmol) and 1-phenyl-3-(thiophen-2-yl)-1H-pyrazole-4-carbaldehyde (2) (254 mg, 1 mmol) in EtOH (2 ml), a solution of NaOH (160 mg, 4 mmol) in EtOH (4 ml) was added at room temperature. The reaction mixture was irradiated by microwaves at 100 W for 9–10 min. After consumption of reactants (as indicated by TLC), aqueous NaOH solution (200 mg, 5 mmol in 2 ml of H2O) and 30% H2O2 (3 ml) were added dropwise and irradiation continued for 2–3 min. After completion of the reaction (monitored by TLC), the resulting light-yellow mixture was poured onto crushed ice and neutralized with dilute HCl. The light-yellow solid thus obtained was filtered off, washed with water, and dried. The crude product was purified by column chromatography on silica gel using hexane–EtOAc, 7:3, as eluent and further recrystallized from EtOH to afford the desired products 3a–h as yellow solids, the respective yields are shown in Table 1.
3-Hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1 H -pyrazol-4-yl]-4 H -chromen-4-one (3a). Mp 172–177°C. R f 0.28 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3161 (O–H), 1606 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 7.14–7.17 (1H, m, H Ar); 7.28 (1H, s, OH); 7.37–7.52 (7H, m, H Ar); 7.62–7.65 (1H, m, H Ar); 7.84 (2H, d, J = 8.0, H Ar); 8.25 (1H, d, J = 6.8, H Ar); 8.79 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 99.8; 108.3; 108.8; 114.6; 114.7; 119.5; 120.3; 121.8; 123.0; 123.1; 125.4; 126.2; 126.6; 137.2; 140.4 (C-3'); 140.6 (C–OH); 146.1; 157.2; 163.9 (C-8a); 172.5 (C=O). Mass spectrum, m/z (I rel, %): 387 [M+H]+ (100). Found, %: C 68.42; H 3.68; N 7.29. C22H14N2O3S. Calculated, %: C 68.38; H 3.65; N 7.25.
6-Fluoro-3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1 H pyrazol-4-yl]-4 H -chromen-4-one (3b). Mp 177–179°C. R f 0.28 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3300 (O–H), 1607 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 6.75 (1H, s, OH); 7.14–7.17 (2H, m, H Ar); 7.37 (2H, d, J = 3.0, H Ar); 7.44–7.46 (2H, m, H Ar); 7.49–7.54 (2H, m, H Ar); 7.82–7.89 (3H, m, H Ar); 8.79 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 108.3; 109.0; 114.2; 118.4; 119.3; 119.9; 120.6; 120.8; 121.9; 122.9; 123.3; 125.1; 126.4; 126.6; 126.8; 127.0; 129.5; 129.6; 134.2; 139.5 (C-3'); 140.6 (C–OH); 142.6; 153.5 (C-8a); 160.0 (C=O). Mass spectrum, m/z (I rel, %): 405 [M+H]+ (100). Found, %: C 65.38; H 3.39; N 6.98. C22H13FN2O3S. Calculated, %: C 65.34; H 3.24; N 6.93.
6-Chloro-3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1 H pyrazol-4-yl]-4 H -chromen-4-one (3c). Mp 173–175°C. R f 0.26 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3433 (O–H), 1603 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 6.80 (1H, s, OH); 7.14–7.21 (2H, m, H Ar); 7.35–7.58 (6H, m, H Ar); 7.82 (2H, d, J = 7.8, H Ar); 8.21 (1H, s, H Ar); 8.79 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 108.9; 119.3; 120.6; 122.1; 122.8; 124.3; 125.7; 126.3; 126.4; 128.0; 128.4; 128.6; 129.2; 133.2; 139.8 (C–Cl); 140.4 (C–OH); 141.0 (C-3'); 150.2; 155.4 (C-8a); 168.7 (C=O). Mass spectrum, m/z (I rel, %): 421 [M+H]+ (100). Found, %: C 62.80; H 3.16; N 6.69. C22H13ClN2O3S. Calculated, %: C 62.78; H 3.11; N 6.66.
6-Bromo-3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1 H pyrazol-4-yl]-4 H -chromen-4-one (3d). Mp 170–172°C. R f 0.24 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3277 (O–H), 1605 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 6.80 (1H, s, OH); 7.12–7.16 (2H, m, H Ar); 7.35–7.54 (5H, m, H Ar); 7.68–7.72 (1H, m, H Ar); 7.82 (2H, d, J = 8.1, H Ar); 8.37 (1H, d, J = 2.4, H Ar); 8.79 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 108.7; 108.9; 119.1; 119.3; 119.6; 120.6; 122.0; 122.3; 122.7; 123.0; 124.3; 125.9; 126.3; 126.6; 129.3; 130.0; 131.8; 132.2; 139.7 (C-3'); 140.4 (C–OH); 141.1; 149.2; 155.3 (C-8a); 168.7 (C=O). Mass spectrum, m/z (I rel, %): 467 [M+H]+ (100). Found, %: C 56.82; H 2.85; N 6.07. C22H13BrN2O3S. Calculated, %: C 56.79; H 2.82; N 6.02.
3-Hydroxy-6-methyl-2-[1-phenyl-3-(thiophen-2-yl)-1 H pyrazol-4-yl]-4 H -chromen-4-one (3e). Mp 154–156°C. R f 0.30 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3281 (O–H), 1601 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 2.48 (3H, s, CH3); 7.13–7.19 (2H, m, H Ar, OH); 7.34–7.39 (2H, m, H Ar); 7.42–7.54 (5H, m, H Ar); 7.82 (2H, d, J = 8.1, H Ar); 8.03 (1H, s, H Ar); 8.77 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 21.8 (CH3); 108.2; 108.5; 113.7; 118.6; 118.9; 121.8; 122.2; 122.6; 123.9; 125.4; 125.8; 128.8; 129.3; 139.5 (C-3'); 140.0; 140.7 (C–OH); 149.7; 155.0 (C-8a); 159.2; 168.3 (C=O). Mass spectrum, m/z (I rel, %): 401 [M+H]+ (100). Found, %: C 68.99; H 4.06; N 7.04. C23H16N2O3S. Calculated, %: C 68.98; H 4.03; N 7.00.
6,8-Dichloro-3-hydroxy-2-[1-phenyl-3-(thiophen-2-yl)-1 H -pyrazol-4-yl]-4 H -chromen-4-one (3f). Mp 183–185°C. R f 0.22 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3257 (O–H), 1612 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 7.07–7.09 (1H, m, H Ar); 7.32–7.40 (4H, m, H Ar, OH); 7.49–7.54 (2H, m, H Ar); 7.65 (1H, d, J = 2.4, H Ar); 7.80 (1H, d, J = 8.1, H Ar); 8.13 (2H, d, J = 2.4, H Ar); 8.69 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 108.3; 119.6; 120.0; 121.9; 122.3; 123.2; 124.3; 125.8; 126.4; 127.0; 127.3; 128.8; 129.3; 130.1; 139.4 (C-3'); 140.6 (C–OH); 142.7; 152.3; 155.3 (C-8a); 168.7 (C=O). Mass spectrum, m/z (I rel, %): 457 [M+H]+ (100). Found, %: C 58.08; H 2.68; N 6.18. C22H12Cl2N2O3S. Calculated, %: C 58.03; H 2.66; N 6.15.
8-Chloro-3-hydroxy-7-methyl-2-[1-phenyl-3-(thiophen-2-yl)-1 H -pyrazol-4-yl]-4 H -chromen-4-one (3g). Mp 182–184°C. R f 0.25 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3251 (O–H), 1605 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 2.34 (3H, s, CH3); 6.80 (1H, s, OH); 7.12–7.16 (2H, m, H Ar); 7.35–7.54 (5H, m, H Ar); 7.68–7.72 (1H, m, H Ar); 7.82 (2H, d, J = 8.1, H Ar); 8.37 (1H, d, J = 2.4, H Ar); 8.79 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 22.1 (CH3); 108.6; 119.0; 119.3; 120.6; 122.1; 123.0; 124.3; 125.6; 126.6; 128.2; 129.2; 130.0; 130.3; 131.8; 137.8; 139.9 (C-3'); 140.4 (C–OH); 150.3; 155.4 (C-8a); 168.7 (C=O). Mass spectrum, m/z (I rel, %): 435 [M+H]+ (100). Found, %: C 63.56; H 3.52; N 6.46. C23H15ClN2O3S. Calculated, %: C 63.52; H 3.48; N 6.44.
3-Hydroxy-6-methoxy-2-[1-phenyl-3-(thiophen-2-yl)-1 H -pyrazol-4-yl]-4 H -chromen-4-one (3h). Mp 162–164°C. R f 0.23 (hexane–EtOAc, 3:1). IR spectrum, ν, cm–1: 3245 (O–H), 1607 (C=O). 1H NMR spectrum, δ, ppm (J, Hz): 3.84 (3H, s, OCH3); 7.13–7.19 (2H, m, H Ar, OH); 7.34–7.39 (2H, m, H Ar); 7.42–7.54 (5H, m, H Ar); 7.82 (2H, d, J = 8.1, H Ar); 8.03 (1H, s, H Ar); 8.77 (1H, s, H-5'). 13C NMR spectrum, δ, ppm: 54.9 (OCH3); 108.2; 108.5; 113.7; 119.2; 120.2; 121.8; 122.2; 122.6; 123.9; 125.3; 125.8; 128.8; 129.3; 139.5 (C-3'); 140.0; 140.7 (C–OH); 149.5; 155.0 (C-8a); 159.1 (C–OMe); 168.3 (C=O). Mass spectrum, m/z (I rel, %): 417 [M+H]+ (100). Found, %: C 66.36; H 3.90; N 6.78. C23H16N2O4S. Calculated, %: C 66.33; H 3.87; N 6.73.
Biological activity tests. All synthesized compounds were screened for their antimicrobial activity against two strains of Gram-positive bacteria (Staphylococcus aureus and Bacillus subtilis), two strains of Gram-negative bacteria (Escherichia coli and Pseudomonas aeruginosa), as well as three strains of fungi (Aspergillus niger, Penicillium italicum, and Fusarium oxysporum). Standard antibiotic drugs ampicillin for bacteria and griseofulvin for fungi were used at a concentration of 50 μg/ml for comparison. The biological activities of these compounds were evaluated by the filter paper disc method38 for 50 μg/ml solutions in DMSO. The inhibition zones of microbial growth surrounding the filter paper disc (5 mm) were measured in millimeters at the end of an incubation period of 3 days at 37°C for E. coli and at 28°C for other bacteria and fungi; DMSO alone showed no inhibition zone.
References
Primo, F. T.; Fröhlich, P. E. Acta Farm. Bonaerense 2005, 24, 421.
Riedel, R. Arzneim. Forsch. 1981, 31, 655.
Kumar, V.; Kaur, K.; Gupta, G. K.; Sharma, A. K. Eur. J. Med. Chem. 2013, 69, 735.
Nargund, L. V. G.; Hariprasad, V.; Reddy, G. R. N. Indian J. Pharm. Sci. 1993, 55, 1.
Sangani, C. B.; Makawana, J. A.; Zhang, X.; Teraiya, S. B.; Lin, L.; Zhu, H.-L. Eur. J. Med. Chem. 2014, 76, 549.
Sankappa Rai, U.; Isloor, A. M.; Shetty, P.; Pai, K. S. R.; Fun, H. K. Arabian J. Chem. 2015, 8, 317.
Blair, B.; Fatheree, R. P.; Fleury, M.; Gendron, R.; Hudson, R.; McKinnell, R. M.; Wilson, M. WO Patent 2011005674.
Secci, D.; Bolasco, A.; Chimenti, P.; Carradori, S. Curr. Med. Chem. 2011, 18, 5114.
Xu, L.; Zhang, X.; Li, X.; Wang, M.; Yuan, B. CN Patent 103232432.
Dong, F.; Chen, X.; Liu, X.; Xu, J.; Li, Y.; Shan, W.; Zheng, Y. J. Chromatogr. A 2012, 1262, 98.
Amr, A.-G.; Abdel-Latif, N. A.; Abdalla, M. M. Acta Pharm. 2006, 56, 203.
Oh, H. C.; Cho, J. H.; El-Gamal, M. KR Patent 2013010514.
Lee, H. I.; Le Hir de Fallois, L. P.; Timmons, P. R.; Cawthorne, W. G.; De Leon, A. P. WO Patent 2008005489.
Sherman, T. D.; Duke, M. V.; Clark, R. D.; Sanders, E. F.; Matsumoto, H.; Duke, O. S. Pestic. Biochem. Physiol. 1991, 40, 236.
Ragab, F. A.; Abdel Gawad, N. M.; Georgey, H. H.; Said, M. F. Eur. J. Med. Chem. 2013, 63, 645.
Mohy El-Din, M. M; Senbel, A. M.; Bistawroos, A. A.; El-Mallah, A.; Nour El-Din, N. A.; Bekhit, A. A.; Abd El Razik, H. A. Basic Clin. Pharmacol. Toxicol. 2011, 108, 263.
Hantoon, M. A. Minn. Med. 2001, 84, 102.
Zhang, X.; Li, X.; Allan, G. F.; Sbriscia, T.; Linton, O.; Lundeen, S. G.; Sui, Z. J. Med. Chem. 2007, 50, 3857.
Yang, X.; Jin, Y.; Liu, H.; Jiang, Y.; Fu, H. RSC Adv. 2012, 2, 11061.
Fong, T. M.; Heymsfield, S. B. Int. J. Obes. 2009, 33, 947.
Rademacher, P. M.; Woods, C. M.; Huang, Q.; Szklarz, G. D.; Nelson, S. D. Chem. Res. Toxicol. 2012, 25, 895.
Bhuiyan, M. H.; Khandkar, M. M. Pak. J. Sci. Ind. Res. 2009, 52, 180.
Murray, M. T. Encyclopedia of Nutritional Supplements; Random House: New York, 1996, p. 320.
Cimanga, K.; Ying, L.; De Bruyne, T.; Apers, S.; Cos, P.; Hermans, N.; Bakana, P.; Tona, L.; Kambu, K.; Kalenda, D. T.; Pieters, L.; Vanden Berghe, D.; Vlietinck, A. J. J. Pharm. Pharmacol. 2001, 53, 757.
Bandgar, B. P.; Patil, S. A.; Korbad, B. L.; Biradar, S. C.; Nile, S. N.; Khobragade, C. N. Eur. J. Med. Chem. 2010, 45, 3223.
Ercelen, S.; Klymchenko, A. S.; Demchenko, A. P. Anal. Chim. Acta 2002, 464, 273.
Klymchenko, A. S.; Avilov, S. V.; Demchenko, A. P. Anal. Biochem. 2004, 329, 43.
Franke, A. A.; Cooney, R. V.; Custer, L. J.; Mordan, L. J.; Tanaka, Y. Adv. Exp. Med. Biol. 1998, 439, 237.
Zwergel, C.; Valente, S.; Salvato, A.; Xu, Z.; Talhi, O.; Mai, A.; Silva, A.; Altucci, L.; Kirsch, G. Med. Chem. Commun. 2013, 4, 1571.
Chohan, Z. H.; Rauf, A.; Naseer, M. M.; Somra, M. A.; Supuran, C. T. J. Enzyme Inhib. Med. Chem. 2006, 21, 173.
Kumar, S.; Pandey, A. K. Sci. World J. 2013, 29, 162750.
Jain, A. C.; Gupta, S. M.; Sharma, A. Bull. Chem. Soc. Jpn. 1983, 56, 1267.
Girish, D. H.; Ashish, P. K.; Atish, H. R.; Rajesh, H. T.; Satish, S. B.; Mahendra, J. P.; Vandana, M. K. Med. Chem. Res. 2014, 23, 461.
Ashok, D.; Vijaya Lakshmi, B.; Ravi, S.; Ganesh, A. Med. Chem. Res. 2015, 24, 1487.
Serdiuk, I. E.; Roshal, A. D.; Błażejowski, J. Chem. Heterocycl. Compd. 2014, 50, 396. [Khim. Geterotsikl. Soedin. 2014, 431.]
Ashok, D.; Ravi, S.; Vijaya Lakshmi, B.; Ganesh, A. J. Serb. Chem. Soc. 2015, 80, 305.
Kiyani, H.; Albooyeh, F.; Fallahnezhad, S. J. Mol. Struct. 2015, 1091, 163.
Saha, K.; Mukherjee Pulok, K.; Mandal, S. C.; Pal, M.; Saha, B. P. Indian Drugs 1995, 32, 402.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary information file to this article containing IR, 1H, 13C NMR and mass spectra of the synthesized compounds is available online at http://springerlink.bibliotecabuap.elogim.com/journal/10593.
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2016, 52(3), 172–176
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 2395 kb)
Rights and permissions
About this article

Cite this article
Ashok, D., Kifah, M.A., Lakshmi, B.V. et al. Microwave-assisted one-pot synthesis of some new flavonols by modified Algar–Flynn–Oyamada reaction and their antimicrobial activity. Chem Heterocycl Comp 52, 172–176 (2016). https://doi.org/10.1007/s10593-016-1852-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-016-1852-4