
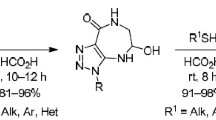
A convenient method was developed for the synthesis of 6-(1H-1,2,3-triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepines, using deoxygenation of 1-(5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazoles with triethyl phosphite.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Deoxygenation of nitroaromatic compounds in the presence of organic trivalent phosphorus derivatives led to the formation of a wide range of nitrogen-containing heterocycles [1–4]. This stepwise reaction involved the formation of nitrene [5–8] and, in the cases when a nucleophilic center was present at the ortho position, led to the formation of condensed heterocyclic system. Both five-membered and six-membered nitrogen-containing heterocyclic systems have been constructed by such approach [9–11]. The lack of a required reactive group at the ortho position might result in benzene ring expansion via enclosure at С–С bond yielding 3H-azepines [12, 13].
Nature of both nitroalkane and reducing agent affects course and scope of the reaction. Several methods are currently known for deoxygenation of primary and secondary nitroalkanes with trivalent phosphorus compounds, leading to the formation of nitriles [14–17], amines [18], and derivatives of oximes [15, 19–21].
There is insufficient information available about the reactivity of tertiary nitro compounds under analogous conditions. Although it is known that the reaction of 2-halo-2-nitropropanes with triethyl phosphite results in phosphate esters of oximes [20], and α-chloronitrocycloalkanes react with triphenylphosphine, giving ring expansion products [22], the deoxygenation of geminal nitro nitroso compounds with triethyl phosphite, on the other hand, occurs at the nitroso group regardless of the substrate structure. This reaction is accompanied by nitrogen atom insertion at the С–N bond with the formation of nitrimines [23].
In this work, for a series of 1-(5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazole derivatives, we studied the possibility of nitro group transformation by the action of triethyl phosphite. The starting compounds were synthesized in three steps from 2-hydroxymethyl-2-nitropropane-1,3-diol (1). In the first step, the nitro alcohol 1 was condensed with dimethoxymethane [24], aldehydes, or ketones [25] in the presence of acidic catalysts, giving compounds 2a,b,c or 2d-g, respectively. The hydroxymethyl derivatives 2a-g were further subjected to oxidative azidation in the presence of K3[Fe(CN)6] under basic conditions, yielding 5-azido-5-nitro-1,3-di-oxanes 3a-g [26].

As shown previously [27], heterocyclic α-nitroazides are highly reactive in 1,3-dipolar cycloaddition to terminal acetylenes under thermal cyclization conditions, giving a mixture of 1,4- and 1,5-triazoles, regardless of the structure of aliphatic heterocycle. Performing the cyclization in the presence of copper(I) salts resulted in selective formation of 1,4-substituted triazoles.
In the current work, we used both 1-substituted and 1,4-disubstituted 1,2,3-triazoles. The mono-substituted triazoles 4a-e were synthesized from the corresponding nitroazides 3a-e and trimethylsilyl acetylene (TMSA). The reaction was performed at room temperature in aqueous methanolic medium in the presence of copper sulfate, ascorbic acid, and potassium carbonate [28]. The process was found to be selective, without the formation of trimethylsilyl-substituted triazoles (according to NMR spectroscopy, LC/MS), irrespective of the structure of 5-azido-5-nitro-1,3-dioxane. The yield of target products practically was not affected by order of the reactant addition, while reaction rate was mostly determined by the solubility of the reactants.

Using the example of equimolar (1:1) mixture of cis- 3e and trans- 3e stereoisomers [26], it was shown that the cycloaddition reaction occurred with equal success for both compounds. The ratio of cis-1-(2-ethyl-2-methyl-5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazole (cis- 4e) and trans-1-(2-ethyl-2-methyl-5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazole (trans- 4e) remained unchanged at 1:1, as determined from NMR spectra, including a NOESY 2D NMR experiment.
The 1,4-disubstituted triazoles 4f-h were obtained by addition of azides 3d,f,g to phenylacetylene in the presence of ascorbic acid and copper(II) sulfate, while 1-(2,2-dimethyl-5-nitro-1,3-dioxan-5-yl)-4-trimethyl-silyl-1H-1,2,3-triazole (4i) was synthesized from azide 3d and TMSA without a catalyst [27].

All triazoles 4a-i were colorless crystalline compounds, which infrared spectra contained bands of asymmetric (1563-1585 cm−1) and symmetric (1332-1380 cm−1) vibrations of nitro group. The intensity of these bands depended on the molecular structure. In comparison, spirodioxanes 4g,h had significantly more intense asymmetric vibration bands and weaker bands of symmetric vibrations.
The character of 1Н NMR signals of dioxane ring methylеne protons (positions 4 and 6) was defined by the presence of methine proton at position 2 of the ring. The spectra of compounds 4a,d-i contained resonance signals due to non-equivalent axial and equatorial protons as two doublets with geminal spin-spin coupling constants of 12.2-13.2 Hz, which is characteristic of substituted 1,3-dioxanes [29]. Each of doublets was transformed into a double triplet in the spectra of compounds 4b,с (2 J = 12.2 Hz, 4 J = 1.6 Hz). The assignment of 13C NMR signals was based on HSQC and HMBC 2D NMR spectra (Table 1). The НМВС spectrum of spirodioxane 4g showed that the 1,4-СН2 and 2,3-СН2 methylene protons of the cyclopentane ring (1.58-1.66 ppm and 1.87-1.94 ppm, respectively), as well as equatorial (4.85 ppm) and axial (5.11 ppm) methylene protons of dioxane ring correlated with the С-5 carbon atom (111.2 ppm). On the other hand, the axial protons of dioxane (5.11 ppm) formed cross peaks with both quaternary carbon atoms С-5 and С-8 (111.2 and 91.2 ppm, respectively). Thus, we conclude that the carbon atom at the dioxane ring position 5 gave a relatively upfield signal (90.4-91.7 ppm) compared to the carbon atom at position 2 of the studied compounds (92.9-111.2 ppm). All the obtained compounds were clearly detected as positive molecular ions [M+H]+ under APCI (100%).
Deoxygenation reaction of 1-(5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazoles 4a-i was studied in benzene with excess of triethyl phosphite (5-10 equiv.). The reaction progress was controlled by TLC and LC/MS. The target products were isolated by column chromatography after the removal of solvent and unreacted triethyl phosphite.

We found that the reaction started at room temperature and was accompanied by selective formation of 4,7-dihydro-1,3,5-dioxazepines 5a-i. The reaction rate was not sensitive to the structure of nitro compounds and was mostly determined by the excess of triethyl phosphite. Increasing the temperature allowed us to shorten reaction time from 35-40 h to 5-15 h, while resulted in substantially lower yields of some target compounds. It was shown by LC/MS in the case of nitrodioxane 4f that deoxygenation at 80-110°С temperature was accompanied by partial degradation, forming 4-phenyl-1,2,3-triazole (6).
The analysis of LC/MS results showed that three compounds were formed during the degradation. Besides the target product 5f (retention time 2.68 min, m/z 273 [M+H]+ (100), 255 [M+H-N2]+ (20)) and 4-phenyl-1,2,3-triazole (6) (retention time 1.82 min, m/z 146 [M+H]+ (100)), we also observed chromatographic peaks due to compounds with retention times 2.57 min (compound 7, m/z 273 [M+H]+ (100)) and 2.62 min (compound 8, m/z 243 (41), 215 (25), 146 (100)). Presence of molecular ion m/z 273 and the lack of nitrogen elimination during ionization and the increased polarity of the compound, as well, suggested the possible formation of 2-substituted 1,2,3-triazole 7 due to migration of dioxazepine substituent at the triazole ring position 2. Similar thermal rearrangements were described in the literature for several 1-alkyl-substituted 1,2,3-triazoles [30–32], while in our hands, thermolysis of nitrodioxane 4f did not result in the formation of 2-substituted 1,2,3-triazole even at 110°С.
The peak with m/z 243 may belong to protonated ion of oxazoline 8, formed by recyclization of dioxazepine ring. Thus, the mechanism leading to 4-phenyl-1,2,3-triazole (6) is not quite obvious. This compound may be formed either directly from dioxazepine 5f or from the products of its transformation.

Thermolysis of dioxazepine 5f showed that 4-phenyl-1,2,3-triazole (6) was formed upon heating in toluene (110°С) even in the absence of phosphorus compounds. The reaction proceeded slowly, with 2-substituted 1,2,3-triazole 7 as the sole by-product.
Deoxygenation reactions in the studied series of compounds were apparently stepwise and occurred analogously to deoxygenation of geminal chloronitrocycloalkanes with triphenylphosphine [22]. The performed study showed that reaction of 1-(5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazoles 4a-i with triethyl phosphite occurred without noticeable heat effect or changes in the color of solution, which was not characteristic to deoxygenation of nitroso compounds [23]. The attempts to detect intermediate 1-(5-nitroso-1,3-dioxan-5-yl)-1H-1,2,3-triazoles 9 by chromatographic methods (LC/MS and TLC) also was not successful. Even with insufficient amount and gradual addition of triethyl phosphite, the target 4,7-dihydro-1,3,5-di-oxazepine 5 was detected immediately.
Thus, the most probable mechanism of dioxane ring expansion was through the stage of ion pair 10, while the formation of 2-substituted 1,2,3-triazole 7 could occur either directly from ion pair 10 or by rearrangement of the target product 5.
The 4,7-dihydro-1,3,5-dioxazepines 5a-i were colorless, crystalline solids. The infrared spectra of these compounds lacked vibrational bands of nitro group, while a strong band appeared in the region of C=N bond vibration (1691-1700 cm−1). The downfield region of 13С NMR spectrum contained resonance signals due to vicinal triazole carbon atoms and another signal at 152-153 ppm. The 1H–13C НМВС spectrum of

spirodioxazepine 5g allowed to establish that the С-9 carbon signal with chemical shift of 152.1 ppm correlated with the signals of 7-СН2 and 10-СН2 methylene protons of 4,7-dihydro-1,3,5-dioxazepine ring (5.40 and 5.19 ppm, respectively). The 1H–15N НМВС spectrum of compound 5g showed correlation of triazole ring proton (9.00 ppm) with all three nitrogen atoms of triazole (264, 353 and 359 ppm), as well as correlation of 4,7-dihydro-1,3,5-dioxazepine ring methylene protons with the nitrogen atom at 288 ppm (Table 2). The obtained results indicate different hybridization of carbon atom adjacent to methylеne groups (CH2O) and the presence of iminium fragment С=N in the structure.
All 4,7-dihydro-1,3,5-dioxazepines 5a-i gave clear mass spectral signals of positively charged ions [M+H]+ (100%) under the conditions of chemical ionization. The presence of phenyl substituent in the triazole ring led to partial extrusion of nitrogen and the appearance of a weak signal (I rel 16-20%) in the mass spectra of products 5f-h, corresponding to the ion [M+H-N2]+.
The structure of these new heterocyclic compounds was finally confirmed by X-ray structural analysis. For compounds 5f,h, the 4,7-dihydro-1,3,5-dioxazepine ring was determined to be in “twist-chair” conformation (Fig. 1, 2), the torsion angle С(2)–N(1)–C(3)–C(4) in both structures was close to 0° (-2.6(5)° in the molecule 5f and -5.87(17)° in the molecule 5h). The C=N bond was practically coplanar to the phenyltriazole fragment: the dihedral angle between triazole ring plane and the plane defined by atoms N(1)–C(3)–N(2) was 5.38(18)° in the molecule 5f and 16.45(10)° in the molecule 5h. At the same time, there was no conjugation between C(3)=N(1) bond and the phenyltriazole ring, because in both cases the C(3)–N(2) bond was ordinary (its length was 1.432(4) Å in the molecule 5f and 1.4333(13) Å in the molecule 5h), while C(3)=N(1) was a double bond (its length was 1.265(4) Å in the molecule 5f and 1.2597(14) Å in the molecule 5h). Analogous situation was observed in the N-imidoylbenzotriazole series [33, 34]. The intermolecular interactions of both compounds in crystalline state were presented by CH···O and CH···N contacts.

Molecular structure of compound 5f with atoms represented by thermal vibration ellipsoids of 50% probability.

Molecular structure of compound 5h with atoms represented by thermal vibration ellipsoids of 50% probability. The methylеne hydrogen atoms at C(13)–C(17) are omitted for clarity.
A few described in the literature 4,7-dihydro-1,3,5-dioxazepines were obtained by thermolysis and photolysis from 1-azirine cycloadducts with diphenyl ketene in 1:2 ratio [35].
Thus, we have demonstrated for the first time the possibility of nitro group deoxygenation in 1-(5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazole derivatives, leading to a series of 6-(1H-1,2,3-triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepine derivatives. The developed method can be used for the preparation of previously unavailable 4,7-dihydro-1,3,5-dioxazepines, enabling characterization of the properties and reactivity of 1,3,5-dioxazepine rings containing a triazole substituent at position 6.
Experimental
IR spectra were recorded on a Thermo Nicolet 360 FTIR instrument in KBr pellets. 1Н and 13С NMR spectra were acquired on a Varian Mercury Plus instrument (400 and 100 MHz, respectively) in DMSO-d6, with TMS as internal standard. The HSQC, 1H–13C HMBC, and 1H–15N HMBC correlation spectra of compounds 4g and 5g were acquired on a Bruker Avance II spectrometer (300 MHz for 1Н nuclei, 76 MHz for 13С nuclei, and 30 MHz for 15N nuclei) in DMSO-d6 at 21°С. The 15N NMR chemical shift values were determined with accuracy to 1 ppm relative to NH3 as external standard. The LC/MS analysis was performed on a Thermo Finnigan Surveyor MSQ with gradient elution and chemical ionization at atmospheric pressure with simultaneous recording of positive and negative ions. Elemental analysis was performed on a Perkin Elmer 2400 Series II CHN-analyzer. Melting points were determined on a Boetius hot stage. Durasil H silica gel (40-63 μm) was used for purification of the obtained compounds by column chromatography.
Compounds 1 [36], 2a [24], 2b [37], 3a-f [26], and 4f,i [27] were synthesized according to published procedures. Phenylacetylene, trimethylsilylacetylene, and triethyl phosphite used in this work were purchased from Sigma-Aldrich.
(5-Nitro-1,3-dioxan-5-yl)methanol (2a) [24]. Yield 63%. Mp 47-48°С (EtOH). IR spectrum, ν, cm−1: 1057 (C–O); 1350, 1544 (NO2); 2782, 2879, 2946, 3007 (C–H), 3451 (O–H). 1H NMR spectrum, δ, ppm (J, Hz): 3.67 (2Н, d, 3 J = 5.6, СН 2ОН); 3.97 (2He, d, 2 J = 12.0) and 4.50 (2Ha, d, 2 J = 12.0, 2OCH2C); 4.70 (1He, d, 2 J = 5.8) and 4.85 (1Ha, d, 2 J = 5.8, OCH2O); 5.44 (1H, t, 3 J = 5.6, CH2OH).
(2-Methyl-5-nitro-1,3-dioxan-5-yl)methanol (2b) [37]. Yield 56%. Mp 67-68°С (EtOH). IR spectrum, ν, cm−1: 1066 (C–O); 1354, 1550 (NO2); 2804, 2901, 2944, 2995 (C–H), 3485 (O–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.15 (3Н, d, 3 J = 5.0, СН 3СН); 3.60 (2Н, d, 3 J = 5.6, СН 2ОН); 3.93 (2He, d, 2 J = 12.8) and 4.58 (2Ha, d, 2 J = 12.8, 2OCH2); 4.74 (1H, q, 3 J = 5.0, CH3СH); 5.37 (1H, t, 3 J = 5.6, CH2OH).
Preparation of Substituted (5-Nitro-1,3-dioxan-5-yl)methanols 2с-g (General Method). Ketone or aldehyde (1.3 mmol) was added to a solution of 2-hydroxymethyl-2-nitropropane-1,3-diol (1) (1.2 mmol) in anhydrous THF (5 ml). The obtained mixture was stirred at room temperature and treated by gradual addition of BF3∙Et2O (1.2 mmol). The solution was maintained for 1-3 h at room temperature (control by TLC). After the reaction was complete, the mixture was quenched with saturated aqueous NaНСO3 (10 ml) and extracted with ethyl acetate (2×10 ml). The organic layer was dried over Na2SO4 and evaporated at reduced pressure. The residue was purified by silica gel column chromatography (1:3 EtOAc–n-hexane). The yields of 5-nitro-1,3-di-oxanes 2c,d [38] were 86 and 73%, respectively.
(2-Ethyl-2-methyl-5-nitro-1,3-dioxan-5-yl)methanol (2e) – mixture of stereoisomers (~1:1, according to 1Н NMR data). Yield 78%. Mp 50-62°С. IR spectrum, ν, cm−1: 1047, 1098 (С–О); 1351, 1547, 1561 (NO2); 2893, 2933, 2950, 2988 (C–H); 3384, 3420 (О–H). cis- Isomer (cis- 2e). 1H NMR spectrum, δ, ppm (J, Hz): 0.77 (3Н, t, 3 J = 7.5, СН 3СН2); 1.36 (3Н, s, СН3); 1.52 (2Н, q, 3 J = 7.5, СН3СН 2); 3.70 (2Н, d, 3 J = 5.7, СН 2OH); 4.07 (2Нe, d, 2 J = 13.2) and 4.37 (2Нa, d, 2 J = 13.2, 2ОСН2); 5.46 (1Н, t, 3 J = 5.7, СН2OH). 13C NMR spectrum, δ, ppm: 7.2 (СН3СН2); 17.5 (СН3С); 32.9 (СН3СН 2); 61.3 (С-4,6); 62.5 (СH2OH); 87.9 (C-5); 99.8 (C-2). trans- Isomer (trans- 2e). 1H NMR spectrum, δ, ppm (J, Hz): 0.82 (3Н, t, 3 J = 7.5, СН 3СН2); 1.20 (3Н, s, СН3); 1.77 (2Н, q, 3 J = 7.5, СН3СН 2); 3.75 (2Н, d, 3 J = 5.7, СН 2OH); 4.02 (2Нe, d, 2 J = 13.2) and 4.31 (2Нa, d, 2 J = 13.2, 2ОСН2); 5.48 (1Н, t, 3 J = 5.7, СН2OH). 13C NMR spectrum, δ, ppm: 8.2 (СН3СН2); 22.8 (СН3С); 25.7 (СН3СН 2); 60.7 (С-4,6); 62.6 (СH2OH); 87.1 (C-5); 100.4 (C-2). Mass spectrum, m/z (I rel,%): 206 [M+H]+ (100) (for both isomers).
(8-Nitro-6,10-dioxaspiro[4.5]dec-8-yl)methanol (2f) . Yield 80%. Mp 101-103°С (CHCl3). IR spectrum, ν, cm−1: 1065, 1107 (С–О); 1339, 1546 (NO2); 2874, 2961 (C–H); 3404, 3345 (О–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.50-1.63 (4H, m, 2CH2); 1.70 (2H, t, 3 J = 7.2, CH2); 1.87 (2H, t, 3 J = 7.2, CH2); 3.70 (2H, d, 3 J = 5.6, CH 2OH); 4.00 (2He, d, 2 J = 13.2) and 4.38 (2Ha, d, 2 J = 13.2, 2OCH2); 5.38 (1H, t, 3 J = 5.6, CH2OH). 13C NMR spectrum, δ, ppm: 20.9 (C-2,3); 29.3 and 34.4 (C-1,4); 60.6 (C-7,9); 62.3 (CH2OH); 87.5 (C-8); 96.9 (C-5). Mass spectrum, m/z (I rel,%): 218 [M+H]+ (100).
(3-Nitro-1,5-dioxaspiro[5.5]undec-3-yl)methanol (2g) . Yield 88%. Mp 95-96°С (EtOH) (mp 96-97°С (decomp.) (MeOH–H2O) [39]). IR spectrum, ν, cm−1: 1056, 1106 (С–О); 1352, 1554 (NO2); 2856, 2934 (C–H); 3403 (О–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.30-1.53 (8H, m, 4CH2); 1.76-1.82 (2H, m, CH2); 3.71 (2H, d, 3 J = 5.4, CH2OH); 4.04 (2He, d, 2 J = 13.2) and 4.33 (2Ha, d, 2 J = 13.2, 2OCH2); 5.36 (1H, t, 3 J = 5.4, CH2OH). 13C NMR spectrum, δ, ppm: 21.9 and 22.0 (C-8,10); 24.8 (C-9); 28.6 and 34.8 (C-7,11); 60.3 (C-2,4); 62.4 (CH2OH); 87.6 (C-3); 98.2 (C-6). Mass spectrum, m/z (I rel,%): 232 [M+H]+ (100).
Preparation of Geminal Azidonitro-1,3-dioxanes 3a-g (General Method). 5-Hydroxymethyl-5-nitro-1,3-dioxane 2a-g (10 mmol) was added to a solution of NaOH (2.00 g, 50 mmol) in water (10 ml). The mixture was stirred at room temperature for 20-30 min; then treated with a solution of NaN3 (3.25 g, 50 mmol) in water (10 ml) and poured into a vigorously stirred solution of K3Fe(CN)6 (16.46 g, 50 mmol) in water (70 ml). The reaction mixture was stirred for 4 h at room temperature and extracted with EtOAc (3×20 ml). The combined extract was washed with water and dried over Na2SO4. The solvent was removed at reduced pressure, the residue was purified by silica gel column chromatography (CH2Cl2). The yields of azidonitrodioxanes 3a-f were 64, 66, 81, 71, 70, and 60%, respectively. The properties of compounds 3a-f matched those reported previously [26].
3-Azido-3-nitro-1,5-dioxaspiro[5.5]undecane (3g). Yield 75%. Colorless oil. IR spectrum, ν, cm-1: 1111 (C–O), 1335, 1557 (NO2), 2124 (N3), 2861, 2939 (C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.34-1.47 (6H, m, 3CH2); 1.66-1.78 (4H, m, 2CH2); 4.16 (2Нe, d, 2 J = 12.8) and 4.46 (2Нa, d, 2 J = 12.8, 2OCH2). 13C NMR spectrum, δ, ppm: 21.9 (С-8,10); 24.6 (С-9); 31.5 and 31.9 (С-7,11); 61.4 (C-2,4); 94.2 (С-3); 99.1 (С-6). Found, %: C 44.65; H 5.81; N 22.87. C9H14N4O4. Calculated, %: C 44.63; H 5.83; N 23.13.
Preparation of 1-(5-Nitro-1,3-dioxan-5-yl)-1 H- 1,2,3-triazoles 4a-i (General Method). А. A solution of K2CO3 (6.25 mmol) and ascorbic acid (1 mmol) in water (10 ml) was stirred at room temperature and treated by adding a solution of trimethylsilylacetylene (6 mmol) in MeOH (20 ml), followed by solution of CuSO4·5H2O (0.5 mmol) in water (10 ml) and then solution of 5-azido-5-nitro-1,3-dioxane 3a-e in MeOH (20 ml). The reaction mixture was stirred for 24 h at room temperature and extracted with CH2Cl2 (3×10 ml). The combined extract was washed with water (20 ml) and dried over Na2SO4. The solvent was removed at reduced pressure, and the residue was purified by silica gel column chromatography, using a 1:1 mixture of EtOAc–n-hexane as mobile phase.
B. A solution of 5-azido-5-nitro-1,3-dioxane 3d,f,g (0.1 mol) and phenylacetylene (0.12 mol) in THF (20 ml) was treated at room temperature by adding a solution of ascorbic acid (0.05 mol) in water (10 ml), followed by solution of CuSO4·5H2O (0.015 mol) in water (5 ml). The reaction mixture was stirred for 2-6 h at room temperature. After the reaction was complete according to TLC, the mixture was diluted with water (20 ml) and extracted with CH2Cl2 (4×20 ml). The organic layer was dried over Na2SO4, the solvent was removed at reduced pressure, and the residue was purified by silica gel column chromatography, using 1:1 mixture of EtOAc–n-hexane as mobile phase.
The yield and properties of triazole 4i matched those reported in the literature [27].
1-(5-Nitro-1,3-dioxan-5-yl)-1 H- 1,2,3-triazole (4a) . Yield 65% (method А). Mp 93-95°С (EtOH). IR spectrum, ν, cm−1: 1154 (С–О); 1332, 1569 (NO2); 2855, 2869, 2933, 2983 (C–H); 3117, 3151 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 4.85 (2He, d, 2 J = 12.4, 2OCH2C); 4.92 (1He, d, 2 J = 6.0) and 5.02 (1Ha, d, 2 J = 6.0, OCH2O); 5.15 (2Ha, d, 2 J = 12.4, 2OCH2C); 7.97 (1Н, d, 3 J = 1.2, H-4 triazole); 8.73 (1Н, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 67.7 (C-4,6); 90.7 (C-5); 92.9 (C-2); 124.7 (C-5 triazole); 134.1 (C-4 triazole). Mass spectrum, m/z (I rel,%): 201 [M+H]+ (100). Found, %: C 36.02; H 4.01; N 27.96. C6H8N4O4. Calculated, %: C 36.00; H 4.03; N 27.99.
1-(2-Methyl-5-nitro-1,3-dioxan-5-yl)-1 H -1,2,3-triazole (4b). Yield 41% (method А). Mp 116-119°С (EtOH). IR spectrum, ν, cm−1: 1113 (C–O); 1337, 1572 (NO2); 2855, 2897, 2928, 2950 (C–H); 3143, 3168 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.14 (3H, d, 3 J = 5.2, CH 3CH); 4.78 (2He, dt, 2 J = 12.2, 4 J = 1.6, 2CH2); 5.07 (1H, q, 3 J = 5.2, CH3CH); 5.23 (2Ha, dt, 2 J = 12.2, 4 J = 1.6, 2CH2); 7.97 (1Н, d, 3 J = 1.2, H-4 triazole); 8.79 (1Н, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 19.5 (СН3); 66.1 (С-4,6); 90.4 (С-5); 99.2 (С-2); 126.0 (С-5 triazole); 134.6 (С-4 triazole). Mass spectrum, m/z (I rel, %): 215 [M+H]+ (100). Found, %: C 39.44; H 4.67; N 26.23. C7H10N4O4. Calculated, %: C 39.25; H 4.71; N 26.16.
1-(5-Nitro-2-phenyl-1,3-dioxan-5-yl)-1 H -1,2,3-triazole (4c). Yield 60% (method А). Mp 150-151°С (EtOH). IR spectrum, ν, cm−1: 1116 (C–O); 1340, 1585 (NO2); 2855, 2887, 2923, 2958 (C–H); 3113, 3151 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 5.04 (2He, dt, 2 J = 12.2, 4 J = 1.6) and 5.42 (2Ha, dt, 2 J = 12.2, 4 J = 1.6, 2CH2); 5.97 (1Н, s, СН); 7.22-7.25 (2Н, m, H Ph); 7.30-7.34 (3Н, m, H Ph); 8.00 (1Н, d, 3 J = 1.2, H-4 triazole); 8.87 (1Н, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 68.5 (С-4,6); 90.6 (С-5); 100.7 (С-2); 125.8 (С-2,6 Ph); 126.0 (С-3,5 Ph); 126.1 (С-5 triazole); 129.1 (С-4 Ph); 134.0 (С-4 triazole); 136.0 (С-1 Ph). Mass spectrum, m/z (I rel, %): 277 [M+H]+ (100). Found, %: C 52.04; H 4.51; N 20.17. C12H12N4O4. Calculated, %: C 52.17; H 4.38; N 20.28.
1-(2,2-Dimethyl-5-nitro-1,3-dioxan-5-yl)-1 H- 1,2,3-triazole (4d). Yield 73% (method А). Mp 104-105°С (EtOH). IR spectrum, ν, cm−1: 1103 (C–O); 1375, 1570 (NO2); 2852, 2922, 2998 (C–H); 3149, 3164 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.39 (3H, s, CH3); 1.42 (3H, s, CH3); 4.80 (2Нe, d, 2 J = 13.2) and 5.06 (2Нa, d, 2 J = 13.2, 2СН2); 7.95 (1Н, d, 3 J = 1.2, H-4 triazole); 8.71 (1Н, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 21.6 (CH3); 24.9 (CH3); 61.9 (С-4,6); 91.3 (С-5); 99.6 (С-2); 124.9 (С-5 triazole); 134.1 (С-4 triazole). Mass spectrum, m/z (I rel, %): 229 [М+Н]+ (100). Found, %: C 42.15; H 5.21; N 24.49. C8H12N4O4. Calculated, %: C 42.10; H 5.30; N 24.55.
1-(2-Ethyl-2-methyl-5-nitro-1,3-dioxan-5-yl)-1 H -1,2,3-triazole (4e) was obtained as 1:1 mixture of stereoisomers, according to 1Н NMR spectral data. Yield 52% (method А). Mp 50-54°С. IR spectrum, ν, cm−1: 1107 (C–O); 1337, 1564 (NO2); 2887, 2936, 2950, 2985 (C–H); 3119 (=C–H). trans- Isomer (trans- 4e). 1H NMR spectrum, δ, ppm (J, Hz): 0.69 (3Н, t, 3 J = 7.6, СН 3СН2); 1.40 (3Н, s, СН3); 1.67 (2Н, q, 3 J = 7.6, СН3СН 2); 4.85 (2Нe, d, 2 J = 12.8) and 5.00 (2Нa, d, 2 J = 12.8, 2ОСН2); 7.95 (1Н, d, 3 J = 1.6, H-4 triazole); 8.73 (1Н, d, 3 J = 1.6, H-5 triazole). 13C NMR spectrum, δ, ppm: 7.1 (СН3СН2); 20.3 (СН3С); 30.9 (СН3 СН2); 61.7 (С-4,6); 91.2 (С-5); 101.0 (С-2); 125.1 (С-5 triazole); 133.9 (С-4 triazole). cis- Isomer (cis- 4e). 1H NMR spectrum, δ, ppm (J, Hz): 0.83 (3Н, t, 3 J = 7.4, СН 3СН2); 1.39 (3Н, s, СН3); 1.70 (2Н, q, 3 J = 7.4, СН3СН 2); 4.76 (2Нe, d, 2 J = 12.8) and 5.10 (2Нa, d, 2 J = 12.8, 2ОСН2); 7.94 (1Н, d, 3 J = 1.6, H-4 triazole); 8.69 (1Н, d, 3 J = 1.6, H-5 triazole). 13C NMR spectrum, δ, ppm: 7.0 (СН3СН2); 18.2 (СН3С); 28.1 (СН3 СН2); 61.6 (С-4,6); 91.2 (С-5); 100.8 (С-2); 124.4 (С-5 triazole); 133.8 (С-4 triazole). Mass spectrum, m/z (I rel, %): 243 [M+H]+ (100) (for both isomers). Found, %: C 44.25; H 5.76; N 23.31. C9H14N4O4. Calculated, %: C 44.63; H 5.83; N 23.13.
1-(2,2-Dimethyl-5-nitro-1,3-dioxan-5-yl)-4-phenyl-1 H- 1,2,3-triazole (4f). Yield 87% (method B). Mp 161-163°С (EtOH) (mp 53-56°С [27] reported for crude product). 1H NMR spectrum, δ, ppm (J, Hz): 1.43 (3H, s, CH3); 1.44 (3H, s, CH3); 4.86 (2Нe, d, 2 J = 12.8) and 5.10 (2Нa, d, 2 J = 12.8, 2СН2); 7.38 (1Н, t, 3 J = 7.2, H-4 Ph); 7.48 (2Н, t, 3 J = 7.6, H-3,5 Ph); 7.90 (2Н, d, 3 J = 7.2, H-2,6 Ph); 9.18 (1Н, s, H-5 triazole). 13C NMR spectrum, δ, ppm: 21.4 (CH3); 24.7 (CH3); 61.8 (С-4,6); 91.6 (С-5); 99.6 (С-2); 120.9 (C-5 triazole); 125.3 (C-2,6 Ph); 128.4 (C-4 Ph); 128.8 (C-3,5 Ph); 129.4 (C-1 Ph); 146.9 (C-4 triazole). Mass spectrum, m/z (I rel, %): 305 [М+Н]+ (100). Found, %: C 55.30; H 5.27; N 18.47. C14H16N4O4. Calculated, %: C 55.26; H 5.30; N 18.41.
1-(8-Nitro-6,10-dioxaspiro[4.5]dec-8-yl)-4-phenyl-1 H -1,2,3-triazole (4g). Yield 82% (method B). Mp 187-189°C (EtOH). IR spectrum, ν, cm−1: 1143 (C–O); 1339, 1564 (NO2); 2855, 2881, 2929, 2968 (C–H); 3106, 3136 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.58-1.66 (4H, m, 2CH2); 1.87-1.94 (4H, m, 2CH2); 4.85 (2He, d, 2 J = 12.8) and 5.11 (2Ha, d, 2 J = 12.8, 2ОCH2); 7.38 (1H, t, 3 J = 7.2, H-4 Ph); 7.48 (2H, t, 3 J = 7.6, H-3,5 Ph); 7.89 (2H, d, 3 J = 7.2, H-2,6 Ph); 9.19 (1H, s, H-5 triazole). 13C NMR spectrum, δ, ppm: 22.5 and 22.9 (C-2,3); 32.8 and 35.2 (C-1,4); 63.4 (C-7,9); 91.2 (С-8); 111.2 (C-5); 120.9 (C-5 triazole); 125.3 (C-2,6 Ph); 128.4 (C-4 Ph); 128.8 (C-3,5 Ph); 129.3 (C-1 Ph); 146.9 (C-4 triazole). Mass spectrum, m/z (I rel, %): 331 [M+H]+ (100). Found, %: C 58.41; H 5.57; N 16.84. C16H18N4O4. Calculated, %: C 58.17; H 5.49; N 16.96.
1-(3-Nitro-1,5-dioxaspiro[5.5]undec-3-yl)-4-phenyl-1 H -1,2,3-triazole (4h). Yield 76% (method B). Mp 167-169°C (EtOH). IR spectrum, ν, cm−1: 1142 (C–O); 1338, 1564 (NO2); 2854, 2885, 2926, 2968 (C–H); 3136 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.37-1.50 (6H, m, 3CH2); 1.72-1.78 (4H, m, 2CH2); 4.86 (2He, d, 2 J = 12.8) and 5.09 (2Ha, d, 2 J = 12.8, 2ОCH2); 7.39 (1H, t, 3 J = 7.2, H-4 Ph); 7.48 (2H, t, 3 J = 7.6, H-3,5 Ph); 7.89 (2H, d, 3 J = 7.2, H-2,6 Ph); 9.17 (1H, s, H-5 triazole). 13C NMR spectrum, δ, ppm: 21.9 (C-8,10); 24.6 (C-9); 29.8 and 33.0 (C-7,11); 61.2 (C-2,4); 91.7 (С-3); 99.5 (C-6); 120.9 (C-5 triazole); 125.3 (C-2,6 Ph); 128.4 (C-4 Ph); 128.8 (C-3,5 Ph); 129.4 (C-1 Ph); 146.9 (C-4 triazole). Mass spectrum, m/z (I rel, %): 345 [M+H]+ (100). Found, %: C 59.32; H 5.94; N 16.14. C17H20N4O4. Calculated, %: C 59.29; H 5.85; N 16.27.
Preparation of 6-(1 H- 1,2,3-Triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepines 5a-i (General Method). A mixture of 1-(5-nitro-1,3-dioxan-5-yl)-1H-1,2,3-triazole 4a-i (0.5 mmol) and triethyl phosphite (3 mmol) in benzene (10 ml) was stirred at reflux. After the completion of reaction (5-15 h, TLC), the solvent was removed by distillation at reduced pressure. The residue was purified by silica gel column chromatography, using 1:5 mixture of EtOAc–n-hexane as mobile phase. The product could be purified as needed by crystallization from ethyl alcohol.
6-(1 H -1,2,3-Triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepine (5a). Yield 41%. Mp 102-105°C (EtOH). IR spectrum, ν, cm−1: 1009, 1097 (C–O); 1694 (C=N); 2906, 2954, 2991 (C–H); 3127, 3164 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 4.99 (2H, s, ОCH2О); 5.25 (2H, t, 5 J = 1.6, СCH2О); 5.41 (2H, t, 5 J = 1.6, NCH2O); 7.88 (1H, d, 3 J = 1.2, H-4 triazole); 8.53 (1H, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 65.2 (C-7); 77.9 (C-4); 95.0 (C-2); 121.9 (C-5 triazole); 133.9 (C-4 triazole); 152.1 (C-6). Mass spectrum, m/z (I rel, %): 169 [M+H]+ (100). Found, %: C 42.95; H 4.91; N 33.12. C6H8N4O2. Calculated, %: C 42.86; H 4.80; N 33.32.
2-Methyl-6-(1 H -1,2,3-triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepine (5b). Yield 72%. Mp 67-70°C (EtOH). IR spectrum, ν, cm−1: 1004, 1119 (C–O); 1699 (C=N); 2915, 2988 (C–H); 3135, 3160 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.34 (3H, d, 3 J = 5.2, CHCH 3); 5.11 (1H, dt, 2 J = 16.8, 5 J = 1.6, СCH2О); 5.24-5.29 (2H, m, СCH2О, CHCH3); 5.33 (1H, dt, 2 J = 16.8, 5 J = 1.4) and 5.51 (1H, dt, 2 J = 14.8, 5 J = 1.6, NCH2O); 7.88 (1H, d, 3 J = 1.2, H-4 triazole); 8.53 (1H, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 18.6 (CH3); 62.8 (C-7); 75.3 (C-4); 99.9 (C-2); 121.9 (C-5 triazole); 133.9 (C-4 triazole); 152.1 (C-6). Mass spectrum, m/z (I rel, %): 183 [M+H]+ (100). Found, %: C 46.35; H 5.62; N 30.41. C7H10N4O2. Calculated, %: C 46.15; H 5.53; N 30.75.
6-(1 H -1,2,3-Triazol-1-yl)-2-phenyl-4,7-dihydro-1,3,5-dioxazepine (5c). Yield 89%. Mp 97-100°C (EtOH). IR spectrum, ν, cm−1: 1012, 1088 (C–O); 1695 (C=N); 2875, 2902, 2925, 2974 (C–H); 3131, 3148 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 5.29 (2H, br. s, СCH2О); 5.44 (1H, dt, 2 J = 14.8, 5 J = 1.6) and 5.49 (1H, dt, 2 J = 14.8, 5 J = 1.6, NCH2O); 6.14 (1H, s, CHPh); 7.35-7.52 (5H, m, Н Ph); 7.91 (1H, d, 3 J = 1.2, H-4 triazole); 8.57 (1H, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 63.2 (C-7); 75.8 (C-4); 100.9 (C-2); 121.9 (C-5 triazole); 126.0 (C-2,6 Ph); 128.2 (C-3,5 Ph); 128.7 (C-4 Ph); 133.9 (C-4 triazole); 137.0 (C-1 Ph); 152.0 (C-6). Mass spectrum, m/z (I rel, %): 245 [M+H]+ (100). Found, %: C 58.94; H 4.88; N 23.05. C12H12N4O2. Calculated, %: C 59.01; H 4.95; N 22.94.
2,2-Dimethyl-6-(1 H -1,2,3-triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepine (5d). Yield 61%. Mp 86-87°C (EtOH). IR spectrum, ν, cm−1: 1009, 1086 (C–O); 1698 (C=N); 2878, 2926, 2944, 2993 (C–H); 3134, 3159 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.44 (6H, s, 2CH3); 5.20 (2H, t, 5 J = 1.8, СCH2О); 5.37 (2H, t, 5 J = 1.8, NCH2O); 7.87 (1H, br. s, H-4 triazole); 8.52 (1H, br. s, H-5 triazole). 13C NMR spectrum, δ, ppm: 23.3 (2CH3); 60.5 (C-7); 73.1 (C-4); 102.4 (C-2); 121.8 (C-5 triazole); 133.8 (C-4 triazole); 151.7 (C-6). Mass spectrum, m/z (I rel, %): 197 [M+H]+ (100). Found, %: C 48.95; H 6.20; N 28.50. C8H12N4O2. Calculated, %: C 48.97; H 6.16; N 28.56.
2-Ethyl-2-methyl-6-(1 H -1,2,3-triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepine (5e). Yield 81%. Mp 65-68°C (EtOH). IR spectrum, ν, cm−1: 1003, 1087 (C–O); 1700 (C=N); 2856, 2883, 2942, 2974 (C–H); 3134, 3164 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 0.88 (3H, t, 3 J = 7.6, CH2CH 3); 1.36 (3H, s, CH3); 1.77 (2H, q, 3 J = 7.6, CH 2CH3); 5.16 (1H, dt, 2 J = 17.2, 5 J = 1.6) and 5.21 (1H, dt, 2 J = 17.2, 5 J = 1.6, СCH2О); 5.34 (1H, dt, 2 J = 15.2, 5 J = 1.6) and 5.39 (1H, dt, 2 J = 15.2, 5 J = 1.6, NCH2O); 7.86 (1H, d, 3 J = 1.2, H-4 triazole); 8.51 (1H, d, 3 J = 1.2, H-5 triazole). 13C NMR spectrum, δ, ppm: 8.1 (CH2 CH3); 19.9 (CH3); 28.1 (CH2CH3); 60.4 (C-7); 73.0 (C-4); 104.5 (C-2); 121.8 (C-5 triazole); 133.8 (C-4 triazole); 151.8 (C-6). Mass spectrum, m/z (I rel, %): 211 [M+H]+ (100). Found, %: C 51.35; H 6.75; N 26.61. C9H14N4O2. Calculated, %: C 51.42; H 6.71; N 26.65.
2,2-Dimethyl-6-(4-phenyl-1 H -1,2,3-triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepine (5f). Yield 37% (63% at reaction temperature of 50°C). Mp 103-105°C (EtOH). IR spectrum, ν, cm−1: 1017, 1084 (C–O); 1701 (C=N); 2888, 2928, 2972, 2988 (C–H); 3164 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.46 (6H, s, 2CH3); 5.23 (2H, t, 5 J = 1.6, СCH2О); 5.42 (2H, t, 5 J = 1.6, NCH2O); 7.36 (1H, t, 3 J = 7.4, H-4 Ph); 7.45 (2H, t, 3 J = 7.6, H-3,5 Ph); 7.96 (2H, d, 3 J = 7.2, H-2,6 Ph); 9.00 (1H, s, H-5 triazole). 13C NMR spectrum, δ, ppm: 23.3 (2CН3); 60.5 (C-7); 73.3 (C-4); 102.5 (C-2); 117.8 (C-5 triazole); 125.5 (C-2,6 Ph); 128.3 (C-4 Ph); 128.9 (C-3,5 Ph); 129.5 (С-1 Ph); 146.6 (C-4 triazole); 151.7 (C-6). Mass spectrum, m/z (I rel, %): 273 [M+H]+ (100), 245 [M+H-N2]+ (18). Found, %: C 61.52; H 5.94; N 20.32. C14H16N4O2. Calculated, %: C 61.75; H 5.92; N 20.58.
9-(4-Phenyl-1 H -1,2,3-triazol-1-yl)-6,11-dioxa-8-azaspiro[4.6]undec-8-ene (5g). Yield 52% (67% at reaction temperature of 50°C). Mp 135-137°C (EtOH). IR spectrum, ν, cm−1: 1008 (C–O); 1691 (C=N); 2851, 2875, 2928, 2957 (C–H); 3142 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.64-1.68 (4H, m, 2CH2); 1.89 (4Н, t, 3 J = 7.0, 2CH2); 5.19 (2H, t, 5 J = 1.6, СCH2О); 5.40 (2H, t, 5 J = 1.6, NCH2O); 7.36 (1H, t, 3 J = 7.4, H-4 Ph); 7.44 (2H, t, 3 J = 7.6, H-3,5 Ph); 7.95 (2H, d, 3 J = 7.2, H-2,6 Ph); 9.00 (1H, s, H-5 triazole). 13C NMR spectrum, δ, ppm: 23.3 (C-2,3); 34.8 (C-1,4); 62.2 (C-10); 75.2 (C-7); 114.7 (C-5); 118.1 (C-5 triazole); 125.6 (C-2,6 Ph); 128.5 (C-4 Ph); 128.9 (C-3,5 Ph); 129.6 (C-1 Ph); 146.8 (C-4 triazole); 152.1 (C-9). 15N NMR spectrum, δ, ppm: 264 (N-1′ triazole); 288 (N-8); 353 (N-3′ triazole); 359 (N-2′ triazole). Mass spectrum, m/z (I rel, %): 299 [M+H]+ (100), 271 [M+H-N2]+ (16). Found, %: C 64.39; H 6.21; N 18.64. C16H18N4O2. Calculated, %: C 64.41; H 6.08; N 18.78.
10-(4-Phenyl-1 H -1,2,3-triazol-1-yl)-7,12-dioxa-9-azaspiro[5.6]dodec-9-ene (5h). Yield 65%. Mp 148- 151°C (EtOH). IR spectrum, ν, cm−1: 1015 (C–O); 1697 (C=N); 2850, 2947 (C–H); 3146 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 1.38-1.53 (6H, m, 3CH2); 1.77 (4H, t, 3 J = 5.8, 2CH2); 5.23 (2H, br. s, СCH2О); 5.42 (2H, br. s, NCH2O); 7.36 (1H, t, 3 J = 7.4, H-4 Ph); 7.45 (2H, t, 3 J = 7.6, H-3,5 Ph); 7.95 (2H, d, 3 J = 7.2, H-2,6 Ph); 8.98 (1H, s, H-5 triazole). 13C NMR spectrum, δ, ppm: 22.4 (C-2,4); 24.6 (C-3); 31.7 (C-1,5); 59.8 (C-11); 72.6 (С-8); 102.4 (С-6); 117.8 (С-5 triazole); 125.5 (C-2,6 Ph); 128.3 (C-4 Ph); 128.7 (C-3,5 Ph); 129.5 (C-1 Ph); 146.6 (С-4 triazole); 151.9 (C-10). Mass spectrum, m/z (I rel, %): 313 [M+H]+ (100), 285 [M+H-N2]+ (20). Found, %: C 65.51; H 6.32; N 17.87. C17H20N4O2. Calculated, %: C 65.37; H 6.45; N 17.94.
2,2-Dimethyl-6-[4-(trimethylsilyl)-1 H -1,2,3-triazol-1-yl]-4,7-dihydro-1,3,5-dioxazepine (5i). Yield 72%. Mp 91-94°C (EtOH). IR spectrum, ν, cm−1: 846 (Si–C); 1022, 1094 (C–O); 1699 (C=N); 2900, 2927, 2960, 2993 (C–H); 3153 (=C–H). 1H NMR spectrum, δ, ppm (J, Hz): 0.27 (9H, s, 3CH3Si); 1.43 (6H, s, 2CH3); 5.18 (2H, br. s, СCH2О); 5.37 (2H, br. s, NCH2O); 8.52 (1H, s, H-5 triazole). 13C NMR spectrum, δ, ppm: -1.48 (Si(CH3)3); 23.3 (2CH3); 60.9 (C-7); 73.2 (C-4); 102.4 (C-2); 127.2 (C-5 triazole); 145.7 (C-4 triazole); 151.7 (C-6). Mass spectrum, m/z (I rel, %): 269 [M+H]+ (100). Found, %: C 49.34; H 7.70; N 20.46. C11H20N4O2Si. Calculated, %: C 49.23; H 7.51; N 20.88.
Thermolysis of 2,2-Dimethyl-6-(4-phenyl-1 H -1,2,3-triazol-1-yl)-4,7-dihydro-1,3,5-dioxazepine (5f). A solution of dioxazepine 5f (272 mg, 1.00 mmol) in toluene (10 ml) was refluxed for 15 h. The solvent was removed at reduced pressure, the residue was separated by silica gel column chromatography, giving 103 mg (71%) of 4-phenyl-1,2,3-triazole (6) as colorless crystals with mp 144-146°C (EtOH) (mp 147-148°C (MeOH–H2O) [40]). IR spectrum, ν, cm−1: 516, 696, 767, 841, 877, 916, 973, 1004, 1084, 1132, 1202, 1230, 1307, 1375, 1454, 1468, 2862, 2963, 3117, 3160. 1H NMR spectrum, δ, ppm (J, Hz): 7.33 (1H, t, 3 J = 7.4, H-4 Ph); 7.44 (2H, t, 3 J = 7.6, H-3,5 Ph); 7.84 (2H, d, 3 J = 7.6, H-2,6 Ph); 8.26 (1H, s, H-5); 14.98 (1H, br. s, NH). 13C NMR spectrum, δ, ppm: 125.4 (C-2,6 Ph); 127.9 (C-4 Ph); 128.7 (C-3,5Ph, С-5); 130.4 (C-1 Ph); 146.7 (C-4). Mass spectrum, m/z (I rel,%): 146 [M+H]+ (100).
X-Ray Sructural Analysis of Compounds 5f,h. Crystals of compound 5f were obtained by slow evaporation of МеОН solution at room temperature and ambient pressure. The crystals were rhombic, space group Pbca; a 9.132(3), b 14.614(5), c 20.428(7) Å; V 2726.0(16) Å3; Z 8 (Z’ 1); d calc 1.327 g∙cm−3; F(000) = 1152. The intensities of 14276 reflections were determined at 100 K temperature on a Bruker APEX II DUO diffractometer (MoKα, λ 0.71073 Å, ω-scanning, 2θ < 53°) and 2780 independent reflections (R int 0.0839) were used in the further refinement.
Crystals of compound 5h were obtained by crystallization from 1:1 mixture of MeOH–2-PrOH. The crystals were monoclinic, P21/n space group; a 5.6366(3), b 14.4327(8), c 18.6719(10) Å; β 91.778(1)°; V 1518.25(14) Å3; Z 4 (Z’ 1); d calc 1.367 g∙cm−3; F(000) 664. The intensities of 19506 reflections were determined at 120 K temperature on a Bruker APEX II diffractometer (MoKα, λ 0.71073 Å, ω-scanning, 2θ < 61°) and 4614 independent reflections (R int 0.0326) were used in the further refinement.
The structures were solved by direct method and sequential synthesis of electron density. Hydrogen atom positions were determined from differential Fourier syntheses. Refinement was performed by F 2 hkl in anisotropic approximation for non-hydrogen atoms. Hydrogen atoms were refined in isotropic approximation according to the “rider” model. Calculations were performed with the SHELXTL 5.10 software suite [41]. The X-ray absorption by the crystal was accounted for by using the SADABS-2008/1 program [42].
The final value of unreliability factors for crystals of compound 5f: R 1 0.0784 (calculated by F hkl for 2081 reflections with I > 2σ(I)), wR 2 0.2144 (calculated by F 2 hkl for all 2780 reflections), the number of refinement parameters 182, GOOF 1.103.
The final value of unreliability factors for crystals of compound 5h: R 1 0.0458 (calculated by F hkl for 3875 reflections with I > 2σ(I)), wR 2 0.1160 (calculated by F 2 hkl for all 4614 reflections), the number of refinement parameters 208, GOOF 1.064.
The complete crystallographic data sets for compounds 5f,h were deposited at the Cambridge Crystallographic Data Center (deposits CCDC 999308 and CCDC 999309, respectively).
References
J. I. G. Cadogan, Synthesis, 11 (1969).
T. Kametani, F. F. Ebetino, T. Yamanaka, and K. Nyu, Heterocycles, 209 (1974).
C. Wentrup, in: A. R. Katritzky (editor), Advances in Heterocyclic Chemistry, Vol. 28, Elsevier, New York (1981), p. 231.
B. C. G. Söderberg, Curr. Org. Chem., 4, 727 (2000).
J. I. G. Cadogan, Acc. Chem. Res., 303 (1972).
G. Smolinsky and B. I. Feuer, J. Org. Chem., 31, 3882 (1966).
T. Kametani, F. F. Ebetino, and K. Fukumoto, Tetrahedron, 30, 2713 (1974).
V. P. Semenov, A. N. Studenikov, and A. A. Potekhin, Chem. Heterocycl. Compd., 14, 233 (1978). [Khim. Geterotsikl. Soedin., 291 (1978).]
V. P. Semenov, A. N. Studenikov, and A. A. Potekhin, Chem. Heterocycl. Compd., 15, 467 (1979). [Khim. Geterotsikl. Soedin., 579 (1979).]
M. R. Devi, J. M. Rao, and V. R. Srinivasan, Synth. Commun., 20, 2301 (1990).
I. V. Ukrainets, L. V. Sidorenko, and O. S. Golovchenko, Chem. Heterocycl. Compd., 43, 1434 (2007). [Khim. Geterotsikl. Soedin., 1687 (2007).]
R. J. Sundberg, B. P. Das, and R. H. Smith, J. Am. Chem. Soc., 91, 658 (1969).
T. de Boer, J. I. G. Cadogan, H. M. McWilliam, and A. G. Rowley, J. Chem. Soc., Perkin Trans. 2, 554 (1975).
S. Trippett and D. M. Walker, J. Chem. Soc., 2976 (1960).
T. Mukaiyama and H. Nambu, J. Org. Chem., 27, 2201 (1962).
P. A. Wehrli and B. Schaer, J. Org. Chem., 42, 3956 (1977).
H. Burgess and J. A. Donnelly, Tetrahedron, 47, 111 (1991).
B. Fischer and L. Sheihet, J. Org. Chem., 63, 393 (1998).
K. S. Kim, E. Y. Hurh, J. N. Youn, and J. I. Park, J. Org. Chem., 64, 9272 (1999).
J. F. Allen, J. Am. Chem. Soc., 79, 3071 (1957).
I. V. Martynov and A. I. Yurtanov, Russ. Chem. Rev., 58, 848 (1989). [Usp. Khim., 1474 (1989).]
M. Ohno and N. Kawabe, Tetrahedron Lett., 7, 3935 (1966).
J. Burdon and A. Ramirez, Tetrahedron, 29, 4195 (1973).
J.-L. Gras, R. Nouguier, and M. Mchich, Tetrahedron Lett., 28, 6601 (1987).
G. B. Linden and M. H. Gold, J. Org. Chem., 21, 1175 (1956).
D. V. Katorov, G. F. Rudakov, and V. F. Zhilin, Russ. Chem. Bull., Int. Ed., 58, 2311 (2009). [Izv. Akad. Nauk, Ser. Khim., 2240 (2009).]
D. V. Katorov, G. F. Rudakov, I. N. Katorova, A. V. Yakushkov, D. P. Simonov, and V. F. Zhilin, Russ. Chem. Bull., Int. Ed., 61, 2114 (2012). [Izv. Akad. Nauk, Ser. Khim., 2098 (2012).]
J. T. Fletcher, S. E. Walz, and M. E. Keeney, Tetrahedron Lett., 49, 7030 (2008).
B. O. Kraiz, Chem. Heterocycl. Compd., 21, 387 (1985). [Khim. Geterotsikl. Soedin., 468 (1985).]
M. Polak and B. Vercek, Synth. Commun., 30, 2863 (2000).
A. Hassner, M. Stern, H. E. Gottlieb, and F. Frolow, J. Org.Chem., 55, 2304 (1990).
P. Magnus, J. Lacour, P. A. Evans, M. B. Roe, and C. Hulme, J. Am. Chem. Soc., 118, 3406 (1996).
K. Niedermann, N. Früh, E. Vinogradova, M. S. Wiehn, A. Moreno, and A. Togni, Angew. Chem., Int. Ed., 50, 1059 (2011).
H.-J. Pi, L.-F. Liu, S.-S. Jiang, W. Du, and W.-P. Deng, Tetrahedron, 66, 6097 (2010).
M. J. Haddadin and A. Hassner, J. Org. Chem., 38, 3466 (1973).
E. Schmidt and R. Wilkendorf, Chem. Ber., 52, 389 (1919).
B. Kedzierski, H. Piotrowska, T. Urbanski, and A. Borys, Rocz. Chem., 46, 1559 (1972).
M. Majewski, D. M. Gleave, and P. Nowak, Can. J. Chem., 73, 1616 (1995).
F. F. Blicke and E. L. Schumann, J. Am. Chem. Soc., 76, 3153 (1954).
Y. Tanaka, S. R. Velen, and S. I. Miller, Tetrahedron, 29, 3271 (1973).
G. M. Sheldrick, Acta Crystallogr., Sec. A: Found. Crystallogr., A64, 112 (2008).
G. M. Sheldrick, SADABS, Program for Scaling and Correction of Area Detector Data, version 2008/1, University of Göttingen, Göttingen (2008).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, No. 11, pp. 1777-1790, November, 2014.
Rights and permissions
About this article

Cite this article
Rudakov, G.F., Dubovis, M.V., Kulagin, A.S. et al. Synthesis of Substituted 6-(1H-1,2,3-Triazol-1-Yl)- 4,7-Dihydro-1,3,5-Dioxazepine. Chem Heterocycl Comp 50, 1634–1646 (2015). https://doi.org/10.1007/s10593-014-1633-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-014-1633-x