
Abstract
Spatial variation in the environment can affect population fitness and individual phenotype by facilitating natural selection and local adaptation, and thereby enhance the diversity and adaptive capacity and persistence of species at regional to continental scales. The song sparrow subspecies complex endemic to the San Francisco Bay region, which has received over a century of close study, presents an opportunity to evaluate the adaptive potential of distinct subspecies faced with habitat loss, population decline, and threats of future environmental change. We used whole-genome sequences from 39 individuals representing five morphologically distinct song sparrow subspecies to evaluate the role of neutral and adaptive evolutionary processes in driving divergence within physiologically challenging habitats across multiple environmental clines. We found that natural selection for traits explained by ecological variables, including temperature and salinity, are drivers of adaptive genetic variation in these song sparrows. Differentiation was highest for candidate loci under selection (compared to neutral markers), as predicted if local ecological processes are at least partially responsible for the rapid radiation of these subspecies. Our findings inform management aimed at conserving and prioritizing population-level diversity in species displaying local adaptation and inhabiting a diverse range of environments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Climate change is occurring at rates likely to exceed the capacity of many populations to adapt via contemporary evolution, casting doubt on their persistence (Anderson et al. 2012). Predicting ‘winners and losers’ in this race remains challenging due to uncertainties about how to characterize the adaptive potential of populations, the effects of historic and contemporary gene flow and natural selection on the pace of local evolution, and the appropriate metrics for prioritizing populations for conservation (Bay et al. 2018; Coates et al. 2018). A growing body of literature aims to define populations’ adaptive capacity, or the ability to cope with environmental change through phenotypic plasticity and evolution (Bay et al. 2017) and to use this information to help inform conservation initiatives. Historically, and in the absence of genetic data, biologists often rely on phenotypic traits to delineate populations for conservation, especially those traits thought to be under genetic control, influential to fitness, or diagnostic of population identity (Haig and Winker 2010). The increasing application of modern genomic methods for systematics, however, provides the opportunity to evaluate adaptive and neutral evolutionary processes, offering additional support for conserving population-level variation (e.g., Oh et al. 2019).
Although these approaches are promising, from a taxonomic perspective, there are concerns that the increased resolution from genomic data may lead to over-splitting if all genetically distinctive populations are classified as full species (Coates et al. 2018; Winker 2021). For birds, most phenotypically distinctive populations have historically been described and formally named as taxonomic subspecies, and in turn these subspecies have often become populations targeted for increased conservation efforts and legal protection (Barrowclough et al. 2016). Some taxonomists, however, have criticized the prioritization of subspecies in conservation, particularly when the subspecies’ classification is based on delineations supported by small numbers of neutral genetic markers, or on morphological traits that are clinal across space (Zink 2004, 2010; Zink and Barrowclough 2008). Such debates arise via the worthy desire to discretize biological diversity, often as a requirement of legal proceedings and legislative policies, versus the contrasting view of speciation and differentiation as an ongoing continuum, a perspective increasingly supported by genomic data (Henderson et al. 2020).
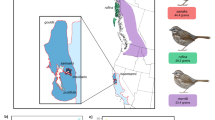
This debate is particularly well developed with respect to a San Francisco Bay complex of five resident song sparrow subspecies: Melospiza melodia heermanni, M. m. maxillaris, M. m. samuelis, M. m. pusillula, and M. m. gouldii (Fig. 1a,c). Song sparrows are widely distributed across North America and display phenotypic variation among their 25 named and 52 described subspecies. The San Francisco Bay region contains the highest concentration of individual subspecies, with five recognized subspecies occupying distinct habitats within a 100 × 70 km region that spans a selective gradient of salt and freshwater habitats. Maxillaris, samuelis, and pusillula are particularly notable for their year-round residency restricted to separate but geographically proximal salt marshes; they occupy a challenging niche that only 25 other vertebrate species have been able to successfully colonize, of which nearly all are of conservation concern due to habitat loss (Greenberg et al. 2006). To this end, subspecies delineations in this system are supported by evidence suggesting rapid adaptation to saline environments (Basham and Mewaldt 1987), differences in coloration (Miller 1956), and genetic isolation (Ferrell 1966), all amounting to substantial evidence of parapatric differentiation (Mayr 1942). In contrast, neither microsatellite (Chan and Arcese 2002) nor mitochondrial DNA (Fry and Zink 1998) data are coincident with these phenotypes (Patten and Pruett 2009). For example, Zink (2004, 2010) concluded that neither subspecies delineations nor neutral genetic markers offer evidence of evolutionary significance or conservation value for these localized populations of sparrows. Most recently, reduced representation genome sequencing provided increased resolution, with subtle separation of the subspecies at microgeographic scales (Mikles et al. 2020). Though the underlying mechanisms of such genomic divergence remain unclear, differentiation is not explained by isolation by distance (Chan and Arcese 2002; Mikles et al. 2020). Here, we use the San Francisco Bay song sparrows as a case study to ask if their subspecies delineations reflect evolutionary processes responsible for the rapid diversification of these populations over the last 10,000 years (Chan and Arcese 2002). If so, then these subspecies classifications would have utility in indicating their evolutionary distinctiveness, and this would in turn imply that the subspecies level of classification has potential merit for recognizing conservation units in other avian taxa.

(A). San Francisco Bay song sparrow ranges digitized. Black dots indicate the sampling locations from the five representative populations and the colors represent the ranges for each subspecies in the Bay (gouldii in red, heermanni in green, samuelis in blue, maxillaris in yellow, and pusillula in orange. (B). Variable importance output ordered by their importance estimated by the RF model. Bio4 = Temperature Seasonality, Bio11 = Mean Temperature of the Coldest Quarter, Bio1 = Annual Mean Temperature, salt = Salinity, bio12 = Annual Precipitation, Bio16 = Precipitation of Wettest Quarter, Bio19 = Precipitation of Coldest Quarter, Bio18 = Precipitation of Warmest Quarter, Bio9 = Mean Temperature of Driest Quarter, Bio10 = Mean Temperature of Warmest Quarter, Bio15 = Precipitation Seasonality, Bio17 = Precipitation of Driest Quarter. (C). Song sparrow subspecies illustrations by Jillian Ditner demonstrate subtle morphological differences among the subspecies. (D). Variation in the top 5 most important variables from RF model by subspecies. Variables were normalized to bring values to range from 0–1. Dark horizontal lines represent the median, colored boxes show the interquartile range, whiskers indicate the 5th – 95th percentile, and dots represent outliers
Methods
Whole genome re-sequencing and variant discovery
We sequenced the genomes of 43 song sparrows representing five subspecies from the San Francisco Bay area (Fig. 1a; Table S1). All birds were sampled during the breeding season (March to May, 1999) by Y. Chan and P. Arcese; adults were captured in mist nets, measured and blood sampled, and released (see Chan and Arcese 2002). We extracted genomic DNA using the DNeasy blood and tissue kit (Qiagen, CA, USA) and quantified DNA concentrations using the Qubit dsDNA High Sensitivity Assay Kit (Life Technologies). Using 200 ng of DNA from each sample, we prepared individually barcoded libraries with a 550 bp insert size following the protocol for the TruSeq Nano DNA Library Prep kit (Illumina, California, USA). We then sequenced libraries for heermanni, maxillaris, and samuelis on a single Illumina NextSeq lane at the Cornell Institute for Biotechnology core facility. We obtained raw sequences for gouldii and pusillula from Walsh et al. (2019a).
We assessed library quality using FastQC version 0.11.8. We used AdapterRemoval version 2.1.1 for sequence trimming, adapter removal, and quality filtering, requiring a minimum Phred quality score of 20 and merged overlapping paired-end reads. We aligned filtered reads to the Song Sparrow reference genome (Feng et al. 2020) using the default settings in BWA 0.7.4 (Li and Durbin 2009) and obtained alignment statistics from Qualimap version 2.2.1 (Okonechnikov et al. 2016). We removed three samples for > 50% missing data, and one for 35% relatedness to another individual (selecting to keep the individual with higher quality mapping statistics). We used Samtools version 1.9 (Li et al. 2009) to convert all resulting BAM files to SAM files and to sort and index files, and Picard Tools version 2.19.2 to add mark duplicates. We used the Haplotype Caller module in GATK version 3.8.1 (McKenna et al. 2010) for single nucleotide polymorphism (SNP) discovery and genotyping and used the following filtering parameters to remove variants: QD < 2, FS > 60.0, MQ < 30.0, and ReadPosRankSum < -8.0. We additionally filtered out variants that were not biallelic, had minor allele frequencies less than 5%, mean coverage less than 2X or more than 50X, and more than 20% missing data. This resulted in a total of 1,630,425 SNPs across all five subspecies.
Characterizing subspecific niche variation
We used existing range maps, bioclimatic variables (WorldClim V2; Fick and Hijmans 2017), and Random Forest (RF; Breiman 2001) to characterize environmental niches for each subspecies to assess possible patterns of ecological divergence in this system (Elith and Leathwick 2009). Briefly, we first georeferenced range maps in Chan and Arcese (2002), Patten et al. (2004), and Patten and Pruett (2009) using georeferencing and editor tools in ArcGIS (version 10.7.1; ESRI 2019) to obtain a contiguous map of the range of each subspecies (Fig. 1a). We note here that our digitized range maps are consistent with maps dating as far back as 1948 (Marshall 1948a) and are an accurate reflection of population ranges through time. To characterize variation in their niches we used a priori knowledge on the effect of climatic conditions and events on song sparrow demography to identify 11 candidate variables: Annual Mean Temperature (bio1), Temperature Seasonality (bio4), Mean Temperature of Driest Quarter (bio9), Mean Temperature of Warmest Quarter (bio10), Mean Temperature of the Coldest Quarter (bio11), Annual Precipitation (bio12), Precipitation Seasonality (bio15), Precipitation of Wettest Quarter (bio16), Precipitation of Driest Quarter (bio17), Precipitation of Warmest Quarter (bio18), Precipitation of Coldest Quarter (bio19) (Table S2) extracted at 30 arc-second spatial resolution from WorldClim, spanning the period of 1970–2000. As an additional candidate variable, we estimated salinity as a proxy using Euclidean distance to the edge of the nearest saline wetland using the spatial analyst tool in ArcGIS and the Areas of Conservation Emphasis Saline Wetlands geospatial data layer, which represents wetland area and location at the HUC12 watershed level (California Department of Fish and Wildlife, 2019). This salinity proxy was determined assuming there is a plateau, and thus was calculated as the Euclidean distance squared, resulting in the further distances being much larger and therefore less affected by salt. Values obtained via our Euclidean distance measurements are an accurate reflection of salinity measurements in the Bay (https://pubs.usgs.gov/of/2017/1022/ofr20171022.pdf), and thus offer a reliable proxy for salinity at our sampling locations.
We used RF to classify and delineate subspecies ranges by their environmental conditions. Within the RF algorithm, trees are produced using a bootstrapped sample of ‘bagged’ data that comprise ~ 64% of all observations, and they are tested against the remaining ‘out-of-bag’ (OOB) data to estimate prediction error (OOB error) as the percentage of incorrectly classified observations. We carried out the tuning and fit of RF in R version 4.0.2 (R Core Team 2020), using the “Caret” and “randomForest” packages (Liaw and Wiener 2002; Kuhn 2020). Our model was trained using five repetitions of a 10-fold cross validation scheme, wherein we randomly split the data into 50% training (n = 30,000) and 50% evaluation data (n = 30,000) to avoid overfitting (Kuhn and Johnson 2013). These data were generated by randomly selecting 60,000 pixels across our study area and associated with subspecies ranges (Fig. 1a). This process generated a final model trained with 11 predictor variables with the best hyperparameter values (mtry = 2; ntree = 500) according to the RMSE parameter (Kuhn and Johnson 2013). To reduce prediction bias, we fit a balanced RF model by growing each tree from random samples of the data with an equal number from each class. We adopted the Gini index to evaluate variable importance, wherein a higher Gini importance indicates a variables’ importance in maintaining predictive power in the RF model. We then evaluated model performance using unseen evaluation data for accuracy, sensitivity, specificity, and Kappa (Kuhn and Johnson 2013). To assess whether the observed subspecies niches were statistically different among the song sparrows, we used an analysis of variance (ANOVA) on the top five most important variables from the final RF model. Variables in this analysis were normalized to bring values to range from 0 to 1 to assess effect size.
Neutral genomic population structure & subspecies delineation
To characterize patterns of genetic structure among subspecies, we ran Admixture version 1.2.3 (Alexander et al. 2009) using a filtered data set (4,961 SNPs) that contained no missing data, was pruned to avoid linkage using the script ldPruning.sh (https://github.com/speciationgenomics/scripts/blob/master/ldPruning.sh), and had putatively adaptive SNPs removed (see below). For this analysis, we investigated five population clusters, using the default settings. For all other analyses of population structure, we assessed patterns and genomic diversity based on the full data set (1,630,425 SNPs) and a putatively neutral SNP data set. To target putatively neutral SNPs, we excluded SNPs that were mapped to exons or intervals within 25 kb of exons (see Kawakami et al. 2014), resulting in 308,973 SNPs. We performed a principal component analysis (PCA) on the full and neutral data sets using the “SNPRelate” package in R (Zheng et al. 2012). For both data sets, we additionally characterized genome-wide patterns of divergence between subspecies by calculating pairwise FST values for each comparison using VCFtools (Danecek et al. 2011). We calculated FST for 25 kb windows across our scaffolds and for individual SNPs, dropping windows with fewer than 10 SNPs. Using pairwise FST estimates for the full data set, we tested for isolation by distance using a Mantel test in R. We quantified genetic diversity by estimating individual heterozygosity and nucleotide diversity in 25 kb windows using VCFtools; for these calculations, we removed all missing data from both datasets, as estimates can be biased by missing data (Schmidt et al. 2021). For the full and neutral data sets, we counted the number of private alleles within each subspecies using bcftools (Li et al. 2009).
Genotype-environment associations
To assess whether the observed genomic differentiation among song sparrows in the San Francisco Bay area was a result of ecological divergence, we scanned for SNPs associated with five environmental variables identified by RF as most important in delineating ecological niches of the subspecies. These variables included temperature seasonality, mean temperature of the coldest quarter, annual mean temperature, salinity, and annual precipitation. We tested for Genotype-Environment Associations (GEA) through a combination of multivariate and univariate approaches: we ran a redundancy analysis (RDA; multivariate) using the rda function in the R package Vegan 2.4-5 (Oksanen et al. 2017), and a latent factor mixed model (LFMM; univariate) using the lfmm function of the LFMM package in R. Both methods are robust to a range of underlying demographic processes and sampling designs (Rellstab et al. 2015; Forester et al. 2018), while providing a balance between error rates and detection power (Carvalho et al. 2020). For both methods, we imputed missing genotypes for the full data set by using the most common genotype at each SNP across all individuals. Because temperature variables were correlated (pairwise Pearson correlation coefficients > 0.7), we ran a PCA on all temperature variables using the prcomp function in R and used the first and second principal components as predictors in the GEA analyses (Frichot et al. 2013). We used results from ADMIXTURE to define the number of latent factors used as K = 3 (Supplemental Material, Figure S1).
RDAs can offer a robust approach to detecting correlations between genotype and environmental data, particularly compared to other differentiation-based outlier scans (Rellstab et al. 2015; Forester et al. 2018). We used an RDA to test for multilocus signatures of selection for multiple environmental variables and evaluated the significance of the RDA using an analysis of variance (ANOVA) with 999 permutations. Loci with a loading ± 4 SD from the mean loading on the first three constrained ordination axes were considered candidates under environmental selection following Forester et al. (2018). We used a Pearson’s correlation (r) to identify environmental variables exhibiting the strongest association with each candidate locus.
For the LFMM analysis, we conducted 5 independent runs of 10,000 iterations and a 5,000 iteration burn-in. We corrected association p-values based on empirical genomic inflation factors (𝝀) to control for false discoveries (François et al. 2016). To do this, we inspected the distribution of p-values to ensure that they followed a normal distribution following François et al. (2016). For all variables, we set 𝝀 = 0.1. When generating a list of candidate SNPs, we used the Benjamini-Hochberg algorithm (Benjamini and Hochberg 1995) with a maximum false discovery rate of 10−5.
Evolutionary distinctiveness and identification of conservation units
Based on the GEA methods above, we identified shared candidate SNPs that correlated with environmental variables, which we refer to from here on as a putatively adaptive SNP data set. To provide a measure of adaptive diversity we calculated pairwise FST and individual heterozygosity with the putatively adaptive SNP data set. To prioritize groups for conservation, we calculated a measure of population distinctiveness for each subspecies. To do this, we calculated the Shapley metric (SH; Volkmann et al. 2014), which can be calculated from unrooted trees. FST estimates from pairwise comparisons of all five subspecies were used to build a NeighborNet network using the neighborNet function in the R package “phangorn” (Schliep 2011; Hudson and Bryant 2006). This network was then used to estimate the genetic contribution of individual tips (Volkmann et al. 2014), with higher SH values indicating higher priority for management.
Results
Whole genome re-sequencing yielded a mean of 16,043,533 reads per individual with the following sample sizes per subspecies: gouldii (n = 10), heermanni (n = 8), samuelis (n = 6), pusillula (n = 9), and maxillaris (n = 6). The mean alignment rate was 97.5%, the mean coverage was 2.60X, and the mean missing data was 12% (Table S1).
Subspecific niche variation
Climate varied predictably across the five subspecies ranges. The top five most influential variables in delineating subspecies niches were temperature seasonality (bio4), mean temperature of the coldest quarter (bio11), annual mean temperature (bio1), annual precipitation (bio12), and distance to saltwater (Fig. 1b). The influence of the top five climate predictors varied significantly among subspecies ranges (Fig. 1d; p < 0.001), except coldest quarter (bio11) and annual mean temperature (bio1) did not differ between the ranges of gouldii (normalized mean ± SD: 0.47 ± 0.17) and maxillaris (0.47 ± 0.05; p > 0.99), and gouldii (0.46 ± 0.19) and samuelis (0.48 ± 0.05; p > 0. 05), respectively. Distance to saltwater over the ranges of the marsh and upland subspecies varied as predicted, given their known ecotypes. Distance to saltwater also varied between the two upland subspecies (gouldii: 0.13 ± 0.18; heermanni: 0.22 ± 0.25; p < 0.001).
The final RF model displayed high accuracy (97.92% ± 0.21), sensitivity (91.81% ± 0.48), and specificity (98.49% ± 0.19), and a mean error rate of 2.27% (± 0.08) and Kappa of 96.75% (± 0.32). Classification error differed among subspecies such that upland subspecies had the lower error rates (gouldii = 2.25%; heermanni = 0.96%) compared to marsh subspecies (maxillaris = 8.58%; pusillula = 15.14%; samuelis = 16.72%).
Neutral genomic Population structure & subspecies delineation
We observed subtle differentiation among the five subspecies. In the full data set, results from Admixture supported K = 1 as the best supported cluster, yet we detected observable structuring up to K = 3. Under this model, clusters corresponded to (1) pusillula, (2) gouldii, and (3) heermanni, maxillaris, and samuelis (Figure S1;a). Based on ~ 1.6 million SNPs, we observed some clustering by subspecies on a PCA (Figure S1;b), with the three salt marsh subspecies separating along axis one (3.92% of variation explained). The most prominent clustering in this PCA was separation of pusillula and samuelis from the rest of the subspecies. This pattern of separation along PC axes was still present but less pronounced for the neutral data set (Figure S2). Genome-wide FST estimates further suggest moderate levels of divergence among San Francisco Bay area song sparrows (Table S3, S4). Mean FST estimates based on the full data set ranged from 0.011 (maxillaris vs. heermanni) to 0.047 (pusillula vs. samuelis; Table S3). Per SNP FST estimates ranged from 0 to 1 in each of the 10 pairwise comparisons. There was no significant difference between FST estimates based on the full data versus FST estimates based on the neutral data set (Paired Sample T-test; df = 9, t = -1.67, p = 0.12). We found no significant correlation between genetic and geographic distance (p = 0.129; Figure S5).
We observed slight differences in heterozygosity across the five subspecies (Figure S3), with pusillula and gouldii populations exhibiting the lowest mean heterozygosity (0.33 and 0.35, respectively). Mean heterozygosity was highest in samuelis (0.46). We observed a significant difference between individual heterozygosity estimates based on the full versus neutral data set (Paired Sample T-test; df = 38, t = -14.7, p < 2.2e-16). Patterns of nucleotide diversity were notably similar among the five subspecies (Figure S4). Based on the full data set, the number of private alleles observed in each subspecies was: 13,247 (0.82%) in gouldii, 11,258 (0.70%) in heermanni, 4,279 (0.29%) in maxillaris, 16,140 (0.98%) in pusillula, and 10,156 (0.62%) in samuelis. The number of private alleles observed in the neutral data set was proportionally comparable: 0.81% in gouldii, 0.88% in heermanni, 0.37% in maxillaris, 1% in pusillula, and 0.78% in samuelis.
Genotype-environment associations
For the RDA, the first three components explained 36.15%, 27%, and 20.4% of the variation, respectively and the full model was significant (p = 0.027). Temperature PC1 showed significant variation with song sparrow genotypes (p = 0.014) and captured approximately 90% of the variation driven by all three temperature variables. Annual precipitation (bio12; p = 0.14), temperature PC2 (p = 0.84), and salinity (p = 0.1) did not show significant variation with genotype. The first two axes of the RDA largely separated pusillula and gouldii from the other three subspecies (Fig. 2a). RDA1 appeared to associate more with salinity and RDA2 associated with the remaining environmental variables. We saw distinct clustering of individuals by subspecies along axes two and three of the RDA, with axis three separating salt marsh populations from upland populations (Fig. 2b). RDA3 appeared to be associated with all the variables, with salinity and annual precipitation being negatively correlated with temperature. Based on our cutoff of ± 4 SD, we identified 171 candidate SNPs that correlated with environmental variables. These included 143 SNPs associated with annual precipitation, 25 SNPs associated with temperature PC1, 1 SNP associated with temperature PC2, and 2 SNPs associated with salinity. Correlations between these candidate SNPs and their most strongly associated environmental variable were moderate, averaging 0.63 (r range = 0.13–0.74).

Subspecies delineation and evolutionary distinctiveness in San Francisco Bay song sparrows. (A) RDA axes 1 vs. 2 and (B) RDA axes 1 vs. 3 support increased divergence among subspecies driven by environmental factors. (C) NeighborNet depicting the relationship among song sparrows. Circle colors correspond to subspecies. Values associated with each tip represent SH metrics for each subspecies, indicating prioritization for management. Legend colors correspond to all panels in this figure
LFMM identified substantially more candidate loci than the RDA. LFMM identified allele frequencies of 11,451 SNPs as significantly associated with environmental variables (282 SNPs for salinity; 8,172 SNPs for annual precipitation; 39 SNPs for temperature PC1; 2,958 SNPs for temperature PC2). Given the discrepancy in the number of outliers identified by LFMM compared to our other methods, we only retained those outliers that were identified by both the RDA and LFMM. For the downstream identification of genes linked to putative regions under selection, we retained 144 SNPs that were identified as outliers by both RDA and LFMM (Table S5).
Evolutionary distinctiveness and identification of conservation units
Mean FST estimates based on the 144 candidate SNPs ranged from 0 (samuelis vs. heermanni and gouldii vs. heermanni) to 0.058 (pusillula vs. samuelis; Table S6). Pairwise FST estimates did not differ significantly between putatively adaptive SNPs compared to estimates based on the full data set (Paired Sample T-test; df = 9, t = -2.56, p = 0.8). The overall pattern of heterozygosity among subspecies based on outlier SNPs was the same as that based on the full data set, with gouldii and pusillula having the lowest per-individual heterozygosity estimates. We observed a significant difference between individual heterozygosity estimates based on the full versus adaptive data set (Paired Sample T-test; df = 38, t = 2.66, p = 0.01). The NeighborNet network for subspecies in the San Francisco Bay is non-tree like, and the close placement of groups to each other on the network is as expected given their recent divergence times (Fig. 2c). We do see the placement of pusillula at a more isolated tip in the network, which is consistent with our other metrics of population structure. The pusillula subspecies had the highest SH rank, as expected based on the network (0.029), followed by maxillaris (0.01). The other subspecies largely clustered together in the network and had lower, and comparable, SH rankings: 0.008 for samuelis, 0.006 for heermanni, and 0.007 for gouldii.
Discussion
An extraordinary amount of attention has been dedicated to the study of the San Francisco Bay song sparrows (Grinnell 1909; Huxley 1942; Grinnell and Miller 1944, 1956, Marshall 1948a,b, Johnston 1956a,b, Mayr 1963, Chan and Arcese 2002, 2003, Mikles et al. 2020). Notable for their high concentration of morphologically distinct subspecies within a small geographic area, these song sparrows present a tractable system for investigating replicated colonization of marsh environments across a habitat gradient variable in salinity, temperature, and precipitation, and one now drastically altered by anthropogenic influence. By combining genomic data with random forest niche modeling, we identified evidence of local selection and putative ecological divergence over a fine spatial scale in the song sparrow subspecies of the San Francisco Bay. We posit that ecological variables linked to microgeographic habitat variation in the Bay are primarily responsible for the rapid radiation of these subspecies. Our work further suggests that selection for adaptive phenotypes, rather than neutral processes linked to drift or divergence time alone, is the primary driver of diversification in this system, however we discuss these conclusions within the context of alternative mechanisms in greater detail below. We conclude that these song sparrows offer lessons for the application of how genomic data can be applied to the characterization and conservation of local genetic diversity.
Evolution and conservation in the San Francisco Bay ecoregion
Saltwater marshes are inherently challenging environments that require specialized adaptations in the vertebrate species that have colonized them (Greenberg et al. 2006). The fragmented and patchy distribution of salt marsh habitats creates further challenges when prioritizing conservation efforts among small populations subject to rapid environmental change. Despite being the largest estuary on the west coast of North America, 90% of the San Francisco Bay marsh habitats have been converted to human use (San Francisco Bay Estuary Project 1991, Takekawa et al. 2006) and those remaining are at risk due to sea level rise in the next century (Thorne et al. 2018). Given the associated conservation challenges, active management of tidal marsh endemics is warranted. By identifying environmental drivers of locally adapted populations in these ecosystems, we can better understand how to preserve the full range of endemic phenotypes represented in the region.
Fine-scale mapping of habitats which appear to be homogenous, such as salt marshes, supported our hypothesis that the ranges of song sparrow subspecies in the San Francisco Bay area varied in microclimate. While we expected variation to be high between salt marsh and upland subspecies ranges, we also observed variation within both salt marsh and upland habitats, consistent with the hypothesis that environmental heterogeneity can facilitate local adaptation at fine spatial scales (Miller 1956; Mayr 1963; Ferrell 1966). Temperatures diverged most between the niches of the two upland subspecies, with gouldii experiencing the coolest conditions on the coast, and heermanni experiencing the warmest in inland habitats. Additionally, salinity varies greatly between upland and salt marsh subspecies but also within the estuary, with the South San Francisco Bay roughly 33 times saltier than the Suisun Bay, which receives large freshwater inputs from the Sacramento and San Joaquin Rivers (Schraga and Cloern 2017). Given ample evidence of rapid adaptation to saline environments in other species of new world sparrows (Walsh et al. 2019a, b, Benham et al. 2020), our discovery of 144 regions that may be associated with an adaptive response to environmental variation is not surprising. However, this is the first use of whole genome data to explore local adaptation among subspecies of song sparrows over a microgeographic scale. Our detailed characterization of covariation in the spatial distributions of habitat and genotype at fine geographic scales offers strong support for the hypothesis that selection has contributed to the rapid diversification of locally adapted types in this system. We acknowledge that despite advances in the approaches to identifying GxE associations, there are several challenges with these methods (Hoban et al. 2016). Significant GEAs can alternatively arise from both neutral population genetic and demographic processes (Hoban et al. 2016). Moreover, linked selection via background selection or hitchhiking can result in increased genomic divergence between populations, with loci correlating strongly with environmental variables by chance (Cruickshank and Hahn 2014). To disentangle these processes, our findings warrant future work with a broader spatial and environmental sampling scheme that can help to develop a robust assessment of local adaptation in song sparrows. However, the lack of strong neutral genetic structure among the subspecies studied here, coupled with the absence of IBD based on multiple marker types (Chan and Arcese 2002; Mikles et al. 2020, this study) offers support for our hypothesis of ecological divergence. Moreover, the identification of outliers associated with candidate genes that have previously been linked to tidal marsh adaptions including HSP90B1 (Wan et al. 2017) and PHF20 (Walsh et al. 2019a) provides compelling candidates for future validation. Despite the above caveats, we feel that our work identifies new and important signals of genetic diversity among these populations, which in turn reflects evolutionary distinctiveness of populations that is potentially beneficial to the persistence of these populations both locally and regionally.
Evolutionary distinctiveness and conservation
Our comparisons of putatively adaptive versus neutral loci suggest a pattern of local adaptation and diversity in the song sparrows that highlights the utility of genomic data sets in resolving population-level patterns of diversification. Our findings show that population differentiation is the highest in San Francisco Bay area song sparrows at putatively adaptive loci. Because all five subspecies were well-delineated over three RDA axes, our findings offer empirical evidence of the value of identifying adaptive variation among populations (Crandall et al. 2000; Fraser and Bernatchez 2001). However, despite broad recognition that assessing genetic distinctiveness is a desirable first step in identifying the appropriate units for conservation (e.g., Funk et al. 2012, Volkmann et al. 2014), the appropriate metrics for assessing genome-wide differentiation remains uncertain (Fernandez-Fournier 2021). Although relatively few empirical studies have focused on the conservation of genomic diversity to date, a growing body of literature describes the potentially complementary roles of adaptive and neutral processes in shaping genomic variation within species and its incorporation in conservation planning (Bonin et al. 2007; Funk et al. 2012). Our results generally support these suggestions by elucidating a system in which the inclusion of genomic data on traits putatively under selection revealed aspects of diversity that could be overlooked given nuanced variation in the phenotypes of the subspecies studied here.
More work is needed to determine how patterns of diversity and distinctiveness may affect the adaptive capacity or evolutionary potential of song sparrow populations at micro-spatial to regional scales. However, given operational definitions of adaptive capacity as the ability a species to cope with environmental change (IPCC 2014), and evolutionary potential as an attribute determining a species’ ability to maintain positive long-term growth rates in novel environmental conditions (Thurman et al. 2020), we suggest each will be maximized by conserving units exhibiting novel variation at loci linked to traits underlying additive genetic variance in individual fitness (Hendry et al. 2018). Specifically, our RDA identified several loci putatively linked to salinity and climate, factors driving selection and local adaptation in a variety of taxa (Kingsolver et al. 2012), consistent with the notion that microgeographic variation in selection can increase divergence at local scales whilst reducing it within populations (Hendry et al. 2018; Funk et al. 2019). In the case of song sparrows, isolation and small effective population size (Mikles et al. 2020) might be expected to increase genetic drift among populations adapted to their contemporary environment but compromise their capacity to accommodate change in the future (Funk et al. 2019). However, because our prior results indicate substantial evidence of contemporary gene flow between the five subspecies studied here (Mikles et al. 2020), it is possible that sufficient admixture currently exists among populations to maintain a capacity to respond to variation in natural selection and environment in future. Given an imperfect understanding of these factors at present, we suggest managers prioritize the conservation of evolutionarily significant units (ESUs) defined broadly, based on morphological, genetic, and/or ecological boundaries likely to reflect underlying adaptive process and maintain genetic variation in fitness (reviewed in Funk et al. 2012). Because phenotypic differentiation at fine spatial scales and in response to ecological gradients known to affect individual fitness is a defining trait of song sparrows in North America (e.g., Aldrich 1984, Arcese et al. 2002, Patten & Pruett 2011), we suggest that conserving morphologically and genetically distinct subspecies across the range is a first step towards conserving resilience and persistence in the species overall. Genomic data, interpreted conservatively, can help reveal adaptive and neutral genetic differentiation that can facilitate the prioritization of cryptic species or populations (Coates et al. 2018). When integrated with diagnosable differences in phenotype, as is the case for San Francisco Bay area song sparrows, whole genome data can help delineate populations with high precision.
Revisiting the Song Sparrow Subspecies of the San Francisco Bay
The five subspecies of song sparrow studied here meet the recommended criteria for consideration as distinct populations (McCormack and Maley 2015) given that they were (1) classified a priori by phenotype, and (2) shown to be differentiated at dozens of loci linked to environmental heterogeneity. We now (3) know that they occupy different environmental niches with respect to climate and salinity. Currently, four of these five subspecies (all but gouldii) are listed as ‘species of special concern’ in California, but all song sparrows in California appear to be declining (Sauer 2020). Our use of whole-genome surveys suggests that focusing on adaptive variation can advance management planning in many widespread species with cryptic underlying differences in genetic traits affecting fitness. Quantifying local evolutionary distinctiveness could facilitate predictions on how the influence of climate change, genetic variation, and natural selection may affect potential rates of local evolution (Garant 2020). Moreover, characterizing local adaptive variation may play a role in informing assisted gene flow among these populations, defined as the managed movement of individuals between populations to mitigate local maladaptation (Kelly et al. 2021).
References
Aldrich JW (1984) Ecogeographical variation in size and proportions of Song Sparrows (Melospiza melodia). Ornithological Monogr 35
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Anderson JT, Inouye DW, McKinney AM, Colautti RI, Mitchell-Olds T (2012) Phenotypic plasticity and adaptive evolution contribute to advancing flowering phenology in response to climate change. Proc Royal Soc B 279:3843–3852
Arcese P, Sogge MK, Marr AB, Patten MA (2002) Song sparow (Melospiza melodia), The Birds of North America Online.In Poole, A. (Ed.). Washington, DC: The Academy of Natural Sciences, Philadelphia, Pennsylvania, and The American Ornithologists’ Union. Retrieved from the Birds of North America Online: http://bna.birds.cornell.edu/bna/species/704
Barrowclough GF, Cracraft J, Klicka J, Zink RM (2016) How many kinds of birds are there and why does it matter? PLoS ONE 11(11):e0166307
Basham MP, Mewaldt LR (1987) Salt water tolerance and the distribution of South San Francisco Bay Song Sparrows. The Condor 89(4):697–709
Bay RA, Rose NH, Logan CA, Palumbi SR (2017) Genomic models predict successful coral adaptation in future ocean warming rates are reduced. Sci Adv 3(11):e1701413
Bay RA, Harrigan RJ, Underwood VL, Gibbs HL, Smith TB, Ruegg K (2018) Genomic signals of selection predict climate-driven population declines in a migratory bird. Science 359:83–86
Benham PM, Cheviron ZA (2020) Population history and the selective landscape shape patterns of osmoregulatory trait divergence in tidal marsh Savannah sparrows (Passerculus sandwichensis). Evolution 74(1):57–72
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc 57(1):289–300
Bonin A, Nicole F, Pompanon F, Miaud C, Taberlet P (2007) Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol 21(3):697–708
Breiman L (2001) Random forests. Mach Learn 45:5–32. https://doi.org/10.1023/A:1010933404324
California Department of Fish and Wildlife (CDFW) (2019) Area of Conservation Emphasis (ACE) II Saline Wetlands by Watershed [GIS data]. CDFW Biogeographic Data Branch. (https://wildlife.ca.gov/Data/Analysis/ACE). Accessed December 23, 2020
Carvalho CS et al (2020) Combining genotype, phenotype, and environmental data to delineate site-adjusted provenance strategies for ecological restoration. Mol Ecol Resour 21:44–58
Chan Y, Arcese P (2002) Subspecific differentiation and conservation of Song Sparrows (Melospiza melodia) in the San Francisco Bay Region inferred by microsatellite loci analysis. Auk. https://doi.org/10.2307/4089964
Chan Y, Arcese P (2003) Morphological and microsatellite differentiation in Melospiza melodia (Aves) at a microgeographic scale. J Evol Biol 16(5):939–947
Coates DJ, Byrne M, Mortiz C (2018) Genetic diversity and conservation units: dealing with the species-population continuum in the age of genomics. Front Ecol Evol 6:165
R Core Team (2020) R: A language and environment for statistical computing. In R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. TREE 15(7):290–295
Cruickshank TE, Hahn MW (2014) Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol 23(13):3133–3157
Danecek P, A., et al (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158
Elith J, Leathwick JR (2009) Species distribution models: ecological explanation and prediction across space and time. Annu Rev Ecol Evol Syst 40:677–697. https://doi.org/10.1146/annurev.ecolsys.110308.120159
ESRI (2019) ArcMap 10.7.1. In ESRI
Feng S et al (2020) Dense sampling of bird diversity increases power of comparative genomics. Nature 587(7833):252–257
Fernandez-Fournier P, Lewthwaite JMM, Mooers A (2021) Do we need to identify adaptive genetic variation when prioritizing populations for conservation? Conserv Genet 22:205–216
Ferrell GT (1966) Variation in blood group frequencies in populations of song sparrows of the San Francisco Bay Region. Evolution 20:369–382
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int J Climatol 37:4302–4315. https://doi.org/10.1002/joc.5086
Forester BR, Lasky JR, Wagner HH, Urban DL (2018) Comparing methods for detecting multilocus adaptation with multivariate genotype-environment associations. Mol Ecol 27(9):2215–2233
François O, Martins H, Caye K, Schoville SD (2016) Controlling false discoveries in genome scans for selection. Mol Ecol 25(2):454–469
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10(12):2741–2752
Frichot E, Schoville SD, Bouchard G, François O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30(7):1687–1699
Fry AJ, Zink RM (1998) Geographic analysis of nucleotide diversity and song sparrow (Aves:Emberizidae) population history. Mol Ecol 7(10):1303–1313
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends in Ecology and Evolution 27(9):489–496
Funk WC, Forester BR, Converse SJ, Darst C, Morey S (2019) Improving conservation policy with genomics: a guide to integrating adaptive potential into U.S. Endangered Species Act decisions for conservation practitioners and geneticists. Conserv Genet 20:115–134
Garant D (2020) Natural and human-induced environmental changes and their effects on adaptive potential of wild animal populations. Evol Appl 13:1117–1127
Greenberg R, Maldonado JE, Droege S, McDonald MV (2006) Tidal marshes: a global perspective on the evolution and conservation of their terrestrial vertebrates. Bioscience 56(8):675–685
Grinnell J (1909) Three new Song Sparrows from California. Univ Calif Publications Zool 5:265–269
Grinnell J, Miller AH (1944) The distribution of the birds of California. Artemesia Press
Haig SM, Winker K (2010) Avian subspecies: Summary and Prospectus. Ornithological Monogr 67:172–175
Henderson EC, Brelsford A (2020) Genomic differentiation across the speciation continuum in three hummingbird species pairs. BMC Evol Biol 20:113
Hendry AP, Schoen DJ, Wolak ME, Reid JM (2018) The contemporary evolution of fitness. Annu Rev Ecol Evol Syst 49:457–476
Hoban S, Kelley JL, Lotterhos KE, Antolin MF, Bradburd G, Lowry DB, …, Whitlock MC (2016) Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am Nat 188(4):379–397
Hudson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23(2):254–267
Huxley J (1942) Evolution, the modern synthesis. Harper and Brothers
Johnston RF (1956a) Population structure in salt marsh song sparrows. Part I. Environment and annual cycle. Condor 58:24–44
Johnston RF (1956b) Population structure in salt marsh song sparrows. Part II: density, age structure, and maintenance. Condor 58:254–271
Kawakami T, Backström N, Burri R, Husby A, Olason P, Rice AM, …, Ellegren H (2014) Estimation of linkage disequilibrium and interspecific gene flow in Ficedula flycatchers by a newly developed 50k single-nucleotide polymorphism array. Mol Ecol Resour 14(6):1248–1260
Kelly E, Owners KT, Rangers CJ, Jolly N, Indigo A, Smart J, Webb, Phillips B (2021) No outbreeding depression in a trial of targeted gene flow in an endangered australian marsupial. Conserv Genet 22:23–33
Kingsolver JG, Diamond SE, Siepielski AM, Carlson SM (2012) Synthetic analyses of phenotypic selection in natural populations: lessons, limitations and future directions. Evolutionary Ecol Evolutionary Ecol 26(5):1101–1118
Kuhn M (2020) Caret: Classification and Regression Training. R Package Version 6.0–79, Version 6.0–86
Kuhn M, Johnson K (2013) Applied predictive modeling. In Applied Predictive Modeling. https://doi.org/10.1007/978-1-4614-6849-3
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1753–1760
Li H, Handsaker B, Wyosoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079and the 1000 Genome Project Data Processing Subgroup
Liaw A, Wiener M (2002) Classification and regression with Random Forest. R News 2:18–22
Marshall JTJ (1948a) Ecologic races of song sparrows in the San Francisco Bay region. Part I Habitat and Abundance Condor 50:193–215
Marshall JTJ (1948b) Ecologic races of song sparrows in the San Francisco Bay region. Part II. Geographic variation. Condor 50:233–256
Mayr E (1942) Systematics and the origin of species. Columbia Univ. Press, New York
Mayr E (1963) Animal species and evolution. Harvard University Press
McCormack JE, Maley JM (2015) Interpreting negative results with taxonomic and conservation implications: another look at the distinctness of coastal California gnatchatchers. Auk 132(2):380–388
McKenna A et al (2010) Genome Res 20(9):1297–1303
Mikles CS, Aguillon SM, Chan YL, Arcese P, Benham PM, Lovette IJ, Walsh J (2020) Genomic differentiation and local adaptation on a microgeographic scale in a resident songbird. Mol Ecol 29:4295–4307
Miller AH (1956) Ecologic factors that accelerate formation of races and species of terrestrial vertebrates. Evolution 10:262–277
Oh KP, Aldridge CL, Forbey JS, Dadabay CY, Oyler-McCance SJ (2019) Conservation genomics in the Sagebrush Sea: population divergence, demographic history, and local adaptation in sage-grouse (Centrocercus spp). Genome Biol Evol 11(7):2023–2034
Okonechnikov KA, Conesa F, García-Alcalde (2016) Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32(2):292–294
Oksanen FJ et al (2017) Vegan: community ecology package. R package version 2.4-3
Patten MA, Pruett CL (2009) The Song Sparrow, Melospiza melodia, as a ring species: patterns of geographic variation, a revision of subspecies, and implications for speciation. Syst Biodivers 7(1):33–62. https://doi.org/10.1017/S1477200008002867
Patten MA, Rotenberry JT, Zuk M (2004) Habitat selection, acoustic adaptation, and the evolution of reproductive isolation. Evolution. https://doi.org/10.1111/j.0014-3820.2004.tb01593.x
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R (2015) A practical guide to environmental association analysis in landscape genomics. Mol Ecol 24(17):4348–4370
San Francisco Estuary Project (SFEP) (1991) San Francisco Estuary Project status and trends report on wetlands and related habitats in the San Francisco Estuary. ABAG Public report to US-EPA. San Francisco Estuary Project, Oakland, CA
Schliep KP (2011) Phangorn: phylogenetic analysis in R Bioinformatics 27(4):592–593
Schmidt TL, Jasper M, Weeks AR, Hoffmann AA (2021) Unbiased population heterozygosity estimates from genome-wide sequence data. Methods Ecol Evol 12(10):1888–1898
Schraga TS, Cloern JE (2017) Water Quality measurements in San Francisco Bay by the U.S. Geological Survey, 1969–2015. Sci Data 4:170098
Takekawa JY et al (2006) Environmental threats to tidal-marsh vertebrates of the San Francisco Bay Estuary. Stud Avian Biology 32:176–197
Thorne KG et al (2018) U.S. Pacific coastal wetland resilience and vulnerability to sea-level rise. Sci Adv 4(2):eaao3270
Thurman LL et al (2020) Persist in place or shift in space? Evaluating the adaptive capacity of species to climate change. Front Ecol Environ 18(9):520–528
Volkmann L, Martyn I, Moulton V, Spillner A, Mooers AO (2014) Prioritizing populations for conservation using phylogenetic networks. PLoS ONE 9(2):e88945
Walsh J et al (2019a) Genomics of rapid ecological divergence and parallel adaptation in four tidal marsh sparrows. Evol Lett 3(4):324–338
Walsh J, Clucas G, MacManes M, Thomas K, Kovach A (2019b) Divergent selection and drift shape the genomes of two avian sister species spanning a saline–freshwater ecotone. Ecol Evol 9(23):13477–13494
Wan Y, Ma C, Wei P, Fang Q, Guo X, Zhou B, Jiang R (2017) Dynamic expression of HSP90B1 mRNA in the hypothalamus of two chinese chicken breeds under heat stress and association analysis with a SNP in Huainan chickens. Czech J Anim Sci 62(2):82–87
Winker K (2021) An overview of speciation and species limits in birds. Ornithology 138(2):ukab006. https://doi.org/10.1093/ornithology/ukab006
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C (2012) Weir. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. https://doi.org/10.1093/bioinformatics/bts606
Zink RM (2004) The role of subspecies in obscuring avian biological diversity and misleading conservation policy. Proceedings of the Royal Society B. 271:561–564
Zink RM, Barrowclough GF (2008) Mitochondrial DNA under siege in avian phylogeography. Mol Ecol 17:2107–2121
Zink RM (2010) Drawbacks with the use of microsatellites in phylogeography: the song sparrow Melospiza melodia as a case study. J Avian Biol 1:1–7
Author information
Authors and Affiliations
Contributions
JW, PA, CM, and IJL conceived and designed the study with input from YC. YC and PA conducted field work and collected samples. CM and JW conducted all laboratory work and CM and JW carried out all bioinformatic analyses. Data analysis and interpretation was conducted by JW, CM, KC, PB, and SA with input from all co-authors. CM wrote the manuscript with input from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mikles, C.S., Arcese, P., Lovette, I.J. et al. Evolutionary divergence and adaptive capacity in morphologically distinct song sparrow subspecies. Conserv Genet 24, 921–932 (2023). https://doi.org/10.1007/s10592-023-01547-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01547-w