
Abstract
In hyper fragmented biomes, conservation of extant biota relies on preservation and proper management of remnants. The maintenance of genetic diversity and functional connectivity in a landscape context is probably key to long-term conservation of remnant populations. We measured the genetic diversity in seedlings and adults of tree Copaifera langsdorffii and evaluated whether edge and density-dependent effects drive natural regeneration in a set of very small and degraded Brazilian Atlantic forest fragments. We evaluated the role of small remnants in the conservation of genetic diversity in a hyper fragmented landscape and discuss the challenge of long-term population sustainability of such altered habitats. High genetic diversity in adults indicated these fragments are valuable targets for C. langsdorffii in situ conservation, but both genetic diversity and divergence among patches decreased in seedlings. In our landscape, regeneration increased as it neared edges and adults; suggesting this population is resilient to fragmentation. However, at a broader scale, current levels of gene flow have not been sufficient to prevent the loss of genetic diversity across generations. Restoration plans, even at a small scale, are necessary to promote fragment connectivity and spatially expand opportunities for the fairly restricted gene flow observed in this severely fragmented Brazilian Atlantic forest region.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Deforestation has extirpated more than a third of worldwide forest cover, leading the remaining forests highly fragmented (Haddad et al. 2015). Forest fragmentation reduces the area available to existing biota and promotes spatial isolation of remaining populations, leading species to an increased risk of local extinction (Saunders et al. 1991; Turner 1996; Young et al. 1996). One potential consequence of habitat fragmentation is erosion of genetic diversity as a result of genetic drift in small populations and inbreeding depression, due to both smaller population sizes and limited migration among remnants (Templeton et al. 1990; Young et al. 1996). In tree species, however, these theoretical expectations have not always been confirmed by empirical studies (Bacles and Jump 2011; Kramer et al. 2008; Lowe et al. 2005). Trees are hypothesized to be highly resilient to genetic erosion due to their high levels of genetic diversity, long generation times, and long-distance pollen dispersal (Hamrick 2004; Sork and Smouse 2006). However, contrasting results in the literature (Aguilar et al. 2008; Kramer et al. 2008; Lowe et al. 2005) highlight that the interplay of habitat loss and degradation and spatial isolation affects tree species differentially. Ecological degradation that usually follows fragmentation disrupts critical biological interactions, with some species more adaptable to disturbed forests than others (Tabarelli et al. 2010). Indeed, the dynamics of such disturbed forests are amplified in comparison to a continuous pristine habitat (Laurance 2002). Then, the outcome on genetic diversity of a particular species may be unpredictable based solely on population genetics theory.
In hyper fragmented biomes, conservation of extant biota relies on maintenance and proper management of forest remnants. From a landscape perspective, even small and severely altered remnants have their value as they still maintain local levels of genetic diversity (Finger et al. 2012; Ganzhorn et al. 2015; Ribeiro et al. 2005; White et al. 2002) and enhance functional connectivity (Ribeiro et al. 2009; Villard et al. 2014), contributing to conservation of genetic diversity on a landscape scale. Measuring this value is challenging because the common approach to study genetic consequences of forest fragmentation is to compare levels of genetic diversity in adults between a continuous forest and remnants (Aguilar et al. 2008), the former acting as a control. When large, contiguous forests are not available for one to compare, it is difficult to identify which fragments have a practical value for conservation and to determine how they should be managed to maintain their value over time (Saunders et al. 1991), and how to promote landscape connectivity in order to ensure long term conservation. A practical approach to identify conservation value of small fragments is application of the concepts of α, β, and γ diversity introduced by Whittaker (1972). He proposed these diversity measures to quantify the partition of species diversity across spatial scales. Translation of Whittaker’s measures of diversity in the context of forest fragmentation would be: α as the diversity measure of each fragment, β as the turnover in diversity composition across fragments, and γ as the total diversity in a set of fragments. Scofield et al. (2012) recently deployed the α, β, and γ diversity framework to characterize the compositional variation in seed source diversity across seed pools, with the objective to evaluate whether the foraging behavior of birds influences the genetic diversity of tree species whose seeds they disperse. They also introduced a pairwise measure of divergence, δ, to evaluate the diversity turnover in seed sources across seed pools. Here we applied the α, β, γ, and δ diversity statistics to measure the variation in allelic composition of Copaifera langsdorffii Desf. trees distributed in a set of small fragments in a biodiversity hotspot to ultimately identify the value of these fragments to conservation. Specifically, we sought to evaluate the extent to which very small fragments play a role in harboring genetic diversity and to assess the contribution of individual patches to overall γ diversity.
The Brazilian Atlantic forest (BAF) is among the five most important biodiversity hotspots (Myers et al. 2000). It harbors astonishing levels of species biodiversity and endemism but has lost most of its original area since the 1500 s (Myers et al. 2000; Ribeiro et al. 2009). Nowadays, the forest cover is reduced to about 11.4–16.0 % of its 1-million km2 original area (Ribeiro et al. 2009). The degree of forest loss and fragmentation of BAF varies among regions following the local topography, with forests in flat areas remaining more fragmented than those found in the rough mountains near the Atlantic coast (Tabarelli et al. 2010). As 83.4 % of the remnants are smaller than 50 ha, Ribeiro et al. (2009) simulated the effect of the loss of <50 ha fragments on the average distance among larger remnants and concluded that even these small fragments play an important role in reducing isolation. These small remnants can serve as ecological stepping stones, and depending on their spatial configuration, promote functional connectivity (Villard et al. 2014). However, small and irregularly shaped forest fragments are under enormous influence of edge effects, the generation of different environmental conditions that penetrate into the remnant forest habitat (i.e., increasing the local temperature and wind velocity, and decreasing humidity) (Murcia 1995). These changes may exceed the physiological tolerance of organisms adapted to the more mesic conditions of the forest interior. Moreover, edge effects also may influence plant-animal interactions key to plant regeneration, such as pollination, seed dispersal and predation (Christianini and Oliveira 2013). As a consequence of the pronounced edge-effects and degree of spatial isolation, small fragments are generally highly disturbed (Tabarelli et al. 2010). Drastic changes in species composition on fragmented BAF have been reported for many taxa (Tabarelli et al. 2010), mainly resulting from local extinctions of the most sensitive species and proliferation of disturbance-adapted generalist species. It leads to the impoverishment of biodiversity over time (Pütz et al. 2011; Santos et al. 2008) and a shift in species composition towards an early successional system (Santos et al. 2008; Tabarelli and Lopes 2008).
As genetic degradation seems to lag far behind ecological degradation (Bacles and Jump 2011; Kramer et al. 2008; Lowe et al. 2005), focus on recruitment and the integration of ecological and genetic approaches could be a strategy to identify whether current habitat changes are threatening population persistence or enhancing local abundance in a given species. Genetic diversity in naturally regenerated seedlings provides indirect evidence of pollen and seed dispersal efficiency in a fragmented landscape. Some late successional characteristics of C. langsdorffii such as large seeds, animal-mediated seed dispersal, and shade tolerance are indicative of edge sensitiveness (Tabarelli et al. 2010), suggesting that beyond adult local abundance, seedlings and saplings of this species should not persist in such small and degraded BAF remnants. Since edge effects are among the main drivers of biodiversity change in fragmented landscapes (Laurance et al. 2011) and the area of edge influence is proportionally larger in small fragments (Ewers et al. 2007; Fletcher Jr 2005; Malcolm 1994), we sought to understand factors driving C. langsdorffii natural regeneration in very small fragments and evaluated seedling and sapling survival for 1 year. Finally, we used α, β, and γ diversity approach to compare genetic diversity between seedlings and adult cohorts. We believe that to attain long-term persistence the species would need to successfully reproduce and regenerate in such disturbed landscapes. The combined study of regeneration dynamics and genetic diversity in naturally regenerated seedlings are key approaches to address the medium to long-term capacity of C. lansdorffii to persist in degraded forests.
Thus, we sampled seedlings, saplings and adults of C. langsdorffii in a set of very small and degraded BAF fragments and applied ecological and genetic approaches to answer three main questions: (i) What is the relative contribution of small fragments to the total genetic diversity in a severely fragmented landscape? (ii) Is genetic diversity maintained along successive generations? (iii) Are edge and density-dependent effects driving the abundance of seedlings and saplings of C. langsdorffii in small Atlantic forest fragments?
Methods
Study species
Copaifera langsdorffii is a tree widely distributed in Brazil from the Amazon to the Atlantic forest and to Cerrado gallery forests (Costa 2007). Flowering follows a supra-annual pattern, and occurs during the wet season, while fruiting occurs in the next dry season (Pedroni et al. 2002). Reproductive phenology studies indicate trees >15 cm diameter at breast height (DBH) are potentially reproductive (Pedroni et al. 2002), an observation confirmed by a 2-year survey of flowering phenology in our study site (Karina Martins unpublished data). C. langsdorffii is pollinated by bees such as Apis melifera and Trigona spinipes (Freitas and Oliveira 2002). The species is self-compatible and predominantly outcrosses, average multilocus outcrossing rate was 0.859 in progenies sampled in a continuous forest (Tarazi et al. 2013a). Seeds are dispersed by several species of generalist birds (Motta Junior and Lombardi 1990) and gravity, with secondary dispersal by ants and mammals (Costa 2007; Motta Junior and Lombardi 1990). Copaifera langsdorffii seedling and sapling regeneration is constrained by negative density-dependent effects in patches with increasing adult basal area (Oliveira-Filho et al. 1996). Previous studies on Copaifera langsdorffii genetic diversity in fragments showed it is a high genetic diversity species with moderate levels of inbreeding (Manoel et al. 2012; Martins et al. 2008; Sebbenn et al. 2011; Tarazi et al. 2013b). Gene flow is also compromised in isolated fragments or in the edge of larger fragments (Manoel et al. 2012; Martins et al. 2008; Sebbenn et al. 2011) where selfing rates and inbreeding are higher (Sebbenn et al. 2011; Tarazi et al. 2013a). Studies using paternity (Manoel et al. 2012; Tarazi et al. 2013b) or parentage analyses (Sebbenn et al. 2011; Tarazi et al. 2013b) approaches also indicated that fragmented populations are not isolated, hinting that forest remnants serve as stepping stones.
Study site and sampling
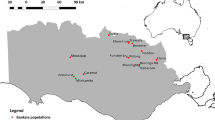
The study was conducted in Atlantic forest fragments in Sorocaba, São Paulo state, in southeastern Brazil, where almost 70 % of the forest remnants are smaller than 20 ha (de Mello et al. 2014). The landscape in our study site is within a transitional ecotone between evergreen and seasonally dry forests, and savannah-like Cerrado vegetation. The forest is highly fragmented, and most forest remnants are <10 ha with the matrix mostly composed of pasture and buildings. Based on a 1962 aerial photograph, forests in our study fragments are secondary, as most fragments were smaller 60 years ago (Corrêa et al. 2014). We censused C. langsdorffii trees >15 cm DBH in a collective group of shredded fragments located in the campus of the Universidade Federal de São Carlos (UFSCar) and the adjacent São Roque farm (hereafter small-fragment, totaling ~15 ha) as well as 1/3 of a larger continuous fragment (~35 ha, hereafter large-fragment) located in Jequitiba farm, approximately 500 m from small-fragment (Fig. 1). Vouchers were collected in UFSCar fragments (Kortz et al. 2014) and deposited in SORO herbarium (#SORO 31). Copaifera langsdorffii is one of the most common species in the study site (Corrêa et al. 2014). We sampled a total of 295 adult trees in small-fragment (~19 trees ha−1) and 339 in large-fragment (~30 trees ha−1). Average DBH was smaller in large-fragment (21.3 cm; CI95 % = 20.7–21.9) than in small-fragment (23.1 cm; CI95 % = 22.4–23.8) (Fig. 1S).

Overview of the study site in a fragmented Atlantic forest landscape in southeastern Brazil. Large-fragment in patch A had 337 trees sampled. Small-fragment was split into four sections. São Roque farm is patch B (n = 105) and the campus area comprehends patches C (n = 94), D (n = 51), and E (n = 44). Seedlings were sampled in two 200 m2 plots located in patches A, C, D and E. Source: Google Earth
Recruitment rate
To evaluate regeneration dynamics we installed 12 plots of 200 m2 in February/March 2013, in small-fragment (10 plots) and in large-fragment (2 plots) and recorded all individuals greater than 30 cm in height. Individuals 30–130 cm in height were considered seedlings, while saplings were taller than 130 cm but ≤ 5 cm in DBH. We resampled the plots in August 2013 and March 2014. Mean annual rate of tree recruitment followed Sheil et al. (1995): \(r = (N_{t} /N_{0} )^{1/t}\), where N t is number of trees at the beginning of study, N 0 is the number of trees at interval end and t is number of years. To correct for differences along the year, we estimated r as the average between the two 6-month intervals. We also evaluated average annual recruitment in number of plants ha−1 year−1 and survival probability for the two size classes (seedlings and saplings).
Seedling and sapling densities
To sample seedling and sapling densities in the entire study area, north–south transects were established at 50 m intervals. We systematically installed 50 m2 circular plots at every 20 m along the transects in small-fragment. In large-fragment, the first two plots closest to the borders were installed every 20 m, but the remaining plots were installed every 100 m. We installed 83 circular plots: 43 in small- and 40 in large-fragments, respectively. In each circular plot, we sampled all C. langsdorffii ≥10 cm height. Since edge effects and negative density-dependent effects often constrain regeneration of tropical plants, we also measured the distance from the center of the plot to the nearest edge and nearest C. langsdorffii adult. To evaluate the influence of distance to the edge (Log) and nearest adult plant (Log) on seedling density, we applied multiple linear regressions. We pooled data for all fragments to increase power. To evaluate how edges and distance to adult plants influenced seed-sapling ratio, we performed another multiple regression of distance to nearest edge (Log) and nearest adult (Log) on the seedling/sapling ratio (square root transformed) in circular plots. Data included only a subsample of circular plots that had at least one seedling or sapling (N = 52).
Laboratory procedures
We sampled either leaves or vascular cambium of all 634 adult trees mapped. We also sampled leaves from each of the 116 mapped seedlings in the 200 m2 plots. All leaves were dried in silica gel and stored in plastic bags at −20 °C. Disks of vascular cambium were stored at −20 °C in 1 mL of transport buffer composed of 0.6 mg of ascorbic acid, 300 ul of CTAB buffer 2 % (Doyle and Doyle 1987), and 700 ul of absolute ethanol. We used a tissue disrupter (SpeedMill Plus, Analytik Jena, Germany) and the innuSPEED Plant DNA Kit (Analytik Jena, Germany) for genomic DNA purification, following manufacturer protocol. Samples were quantified by visual comparison with λ DNA standards after submarine electrophoresis in 1 % agarose gel and Gel Red™ stain (Biotium, USA). Samples were diluted to a final concentration of 2.5 ng μl−1 and were genotyped with eight microsatellite loci (Ciampi et al. 2000). For the polymerase chain reaction (PCR), we used 7.5 ng of DNA, 0.9 μM of each primer and 1× GoTaq Colorless Master Mix (Promega, USA) in a final volume of 15 μL. The PCR protocol consisted of 95 °C for 5 mi; 30 cycles of 94 °C for 1 min, specific annealing temperatures for each primer for 1 min (Martins et al. 2008), and 72 °C by 1 min; followed by a final step at 72 °C for 7 min. PCR fragments were separated through capillary electrophoresis (FS 96 Fragment Analyzer, Advanced Analytical, USA) using the manufacturer protocol (DNF-900 kit, Advanced Analytical, USA). We used the software ProSize 2.0 (Advanced Analytical, USA) for genotyping. We repeated the PCR up to three times for samples that initially failed in order to eliminate missing data from low-quality DNA or human errors. The average frequency of missing data in our data set was 0.8 % (maximum 2.5 % in locus CL20).
Genetic data analysis
We estimated allelic richness (R s ) according to El Mousadik and Petit (1996), expected heterozygosity (H E ), and inbreeding coefficient (F IS ) in adults and seedlings with FSTAT (Goudet 1995). Estimates were compared through non-parametric Kruskal–Wallis test in R (R CoreTeam 2015). Null allele frequencies were estimated following Brookfield (1996), Eq. 3, using MicroChecker (Van Oosterhout et al. 2004). We used a multivariate approach proposed by Jombart et al. (2008) to investigate the spatial pattern of genetic diversity in adult trees. The spatial Principal Component Analysis (sPCA) uses Moran’s I to compare allele frequencies among neighboring sites (spatial autocorrelation), and performs better than PCA because the spatial structure of the sampling area is included in the model from the previous connection network built among individuals. The connection network defines which individuals are neighbors. In animals, distance-based connection networks are defined according to the overlapping of home ranges. In plants, we can use the dispersal distances of pollen and seeds to define neighbor limits. In C. langsdorffii we considered 240 m as the limiting distance to consider two individuals as neighbors. This limit is based on the published maximum pollen and seed dispersal distances estimated with paternity or parentage approaches (Sebbenn et al. 2011; Tarazi et al. 2013b). Monte-Carlo multivariate tests assessed if significant patterns of positive (global) and/or negative (local) spatial correlation existed, as described in Jombart et al. (2008). We used the package ‘adegenet’ (Jombart 2008) in R (R CoreTeam 2015) to perform sPCA analyses.
Because the spatial structure revealed by sPCA may be influenced by the predefined connection network (Demšar et al. 2013), we also used the geo-referenced adult genotypes to identify genetic discontinuities in the adult population using a Bayesian model within a Markov Chain Monte Carlo (MCMC) scheme implemented in Geneland (Guillot et al. 2005a, b). We ran 6 independent repetitions at each K with a run length of 100,000 iterations and a thinning of 100 (each 100th iteration was saved), and k values ranging from 1 to 10. The run giving the highest average posterior probability of K was chosen.
Subsequently, we separated the studied fragments in five sections (Fig. 1), to evaluate the contribution of each patch of small-fragment to the overall genetic diversity in adults and seedlings. We used the allelic α, β, and γ diversity measures of Sork et al. (2015), which follows the statistical framework described in Scofield et al. (2012). The allele diversity within the gth patch was defined as
where \(x_{gk}\) is the number of individuals having the allele (\(k\)) found within patch (\(g\)), and \(n_{g}\) is the total number of individuals sampled in that patch. The average within-patch allele diversity (\(\bar{\alpha }\)) was computed as the unweighted average of the \(\alpha\)-values of all five \(G\) patches. Differences among patches and between seedlings and adults were tested through non-parametric statistics (Mann–Whitney or Kruskal–Wallis tests) in R (R CoreTeam 2015). The total \(\gamma\) diversity was calculated following the equation
where \(X_{k}\) is the total number of trees having the \(k{\text{th}}\) allele across all patches and \(N\) the total number of individuals sampled. \(\beta\) diversity was defined as \(\beta = \left( {\gamma /\alpha } \right)\). We also calculated a [0,1]-scaled measure of allele compositional divergence between the \(g{\text{th}}\) and the \(h{\text{th}}\) patches (\(\delta_{gh}\)):
where \(r_{gh}\) is defined as
We then calculated an average [0,1]-scaled measure of allele divergence (\(\bar{\delta }\)) among patches following the equation below:
We tested whether \(\bar{\delta }\) statistically differed from zero using the permutation procedure described in Scofield et al. (2012). Finally, average overlap (\(\bar{\omega }\)) in allele diversities among patches was obtained following \(\bar{\omega } = 1 - \bar{\delta }\). Computations were executed in R (R CoreTeam 2015) following functions available at https://github.com/douglasgscofield/dispersal, and the R package ‘readGenalex’ available via CRAN (http://cran.r-project.org).
Results
Recruitment rates, seedling and sapling density
The recorded regeneration was 242 seedlings ha−1 year−1 and 2.64 seedlings adult−1 year−1. We estimated seedling survival probability at 84 %. During this period, no seedlings recruited to the sapling class, due to slow growth rates (data not shown) and there was no sapling mortality. Average recruitment rate was 1.31 % (±0.10 %) and considering 95 % confidence intervals around the mean, estimates did not significantly vary from the first (1.48 %, CI 1.10–1.85) to the second semester (1.15 % CI 0.93–1.37), probably because of small number of seeds produced in that season (Karina Martins personal observation). Considering data from the 83 circular plots, we estimated 369 seedlings ha−1 (10 cm < height < 130 cm). The variability among plots was large (SD = 517), as seedlings were only found in 59 % of the plots. We estimated 99 saplings ha−1 (height > 130 cm, DBH < 15 cm) and sapling variability was smaller than in seedlings (SD = 200); saplings were observed in only 30 % of the plots. Distance to the nearest edge and adult plant had weak but significant negative effects on the abundance of seedlings in the circular plots (slopes of –0.50 and –0.30 for distance to the edge and nearest adult, respectively; full model adjusted r 2 = 0.10; df = 2, 85; F = 5.8; p = 0.0043). Seedling/sapling ratio also followed a similar negative relationship with distance to the edge and nearest adult (slopes –0.29 and –0.37, respectively; full model r 2 = 0.14; df = 2, 49; F = 5.31; p = 0.008). These results suggest that C. langsdorffii regeneration increased as it neared edges and adults.
Genetic diversity and structure
Estimates of genetic diversity were high in adults and seedlings (Table 1). Allele richness and expected heterozygosity were slightly lower in seedlings. However, the differences were not significant (H = 4.36, df = 3, p = 0.2253 and H = 4.599, df = 3, p = 0.2036 for \(H_{E}\) and \(R_{S}\) respectively). Null alleles (Table 2) and inbreeding contributed to the high \(F_{IS}\) estimates.
The Monte-Carlo Global test detected a clear spatial pattern of allele frequencies (max(t) = 0.0061, p = 0.002), while the Monte-Carlo Local test was not significant (max(t) = 0.0058, p = 0.16), revealing that close individuals tend to be similar (positive spatial correlation). Three scores were maintained in sPCA (Fig. 2). The first score (I = 0.509) separated trees of small-fragment from large-fragment (Fig. 2a). The second score (I = 0.391) separated small-fragment in two parts, named hereafter São Roque (white squares in Fig. 2b) and campus (the black squares in small-fragment, Fig. 2b) and the third score revealed a weaker structure (I = 0.254) in both small- and large-fragments. The relative contribution of the first three global scores of genetic structure in adults can also be visualized in Fig. 2S. The Bayesian assignment procedure implemented in Geneland yielded similar results as sPCA (results not shown). Adult trees were assigned to three genetic clusters in all the six independent runs while large-fragment was considered one cluster and small-fragment was separated in two clusters, São Roque and campus.

Analyses of multivariate spatial structure of C. langsdorffii adult populations in a fragmented Atlantic forest landscape in Brazil. First (a), second (b) and third (c) principal components of sPCA
Average unweighted within-patch diversity was \(\bar{\alpha }\) = 11.78 (SE ± 0.58, g = 5 patches). All the five patches (white boxes in Fig. 3) had similar levels of alpha diversity (H = 4.35, df = 4, p = 0.3604). The total diversity was γ = 13.1 which did not differ from \(\bar{\alpha }\), meaning that the addition of each small-fragment patch results in a relatively small contribution to the total γ diversity (Fig. 3S). The turnover in allelic composition from patch to patch was β = 1.16, which is the numerical equivalent of “effective number” of patches. The average degree of allele divergence among the five patches was low \(\bar{\delta }\) = 0.197 but differed significantly from the null hypothesis of no divergence (p = 1 × 10−4). The overall overlap in allele composition across patches was \(\bar{\omega }\) = 0.803. Despites the variation among loci, pairwise δ was generally smaller when patches in small-fragment were compared; A–D, C–D, and D–E comparisons yielded the smallest divergences (Fig. 4).

Alpha diversity averaged across loci in adults (Ad) and seedlings (Sd) of C. langsdorffii in the large-fragment (patch A) and in four patches (B, C, D, and E) of small-fragment in an Atlantic forest landscape. Seedlings were not sampled in patch B

Pairwise δ averaged across loci calculated for adults C. langsdorffii in large-fragment (patch A) and small-fragment (patches B, C, D, and E) in an Atlantic forest landscape
Average alpha-diversity was smaller in seedlings than in adults (W = 706, p = 0.0094, Table 3). Within-patch alpha diversity tended to be lower in seedlings than in adults in all four patches compared. However, due to the variation across loci, the smaller within-patch alpha diversity in seedlings was significant only in patch D (W = 55, p = 0.0148) (Fig. 3). Genetic similarity among patches were higher in seedlings than in adults. Average pairwise divergence in seedlings did not differ from the null expectation (Table 3).
Discussion
In spite the small size of the studied fragments and the significant time elapsed since fragmentation (more than 60 years), levels of genetic diversity were slightly greater than those reported in other adult C. langsdorffii populations studied in BAF (Manoel et al. 2012; Martins et al. 2008; Sebbenn et al. 2011; Tarazi et al. 2013b). As most of these comparison studies also sampled fragmented and small populations (except Tarazi et al. 2013b), our greater genetic diversity can be explained by existing pre-fragmentation genetic diversity (Hamrick 2004) and high local abundance of trees in our study sites (Ganzhorn et al. 2015), which prevented the pervasive effects of genetic drift. The loss of genetic variability due to fragmentation is more difficult to detect in long-lived tree species. Genetic erosion is usually only detected in older remnants (>100 years), due to cumulative effects of genetic drift through several generations, and/or after a severe demographic bottleneck (Aguilar et al. 2008). We found greater inbreeding coefficient estimates than previous studies that used the same markers (Sebbenn et al. 2011; Tarazi et al. 2013b). Although the presence of null alleles (Table 2) and genotyping errors could have contributed to these large estimates of \(F_{IS}\), similar high levels of inbreeding were documented in other C. langsdorffii studies as well (Manoel et al. 2012; Martins et al. 2008). Observed levels of inbreeding are probably a consequence of high selfing rates (Tarazi et al. 2013a), which resulted in small estimates of observed heterozygosity; low inbreeding depression (Tarazi et al. 2013b), contributing to keep inbred individuals in these populations; and Wahlund effect (Sebbenn et al. 2011). As we used an indirect method to detect null alleles, based on the comparison between observed and expected heterozygosities under Hardy–Weinberg Equilibrium (HWE), we believe null allele estimates are overestimated. Dabrowski et al. (2014) and Dabrowski et al. (2015) argue that there is no totally reliable method to estimate null alleles in natural populations, because HWE is rarely met in such populations. Considering that actual null allele frequencies are usually unknown, error rates in null allele frequencies are difficult to estimate (Dabrowski et al. 2014), reinforcing the belief that these estimates are imprecise. Several characteristics of C. langsdorffii reproductive biology support our hypothesis regarding inbreeding in this population. The species is self-compatible and mixed mated (Tarazi et al. 2013a), flowering is supra-annual, and a small percentage of trees flowers each reproductive season (Pedroni et al. 2002). We also observed this flowering pattern in our study site (Karina Martins, unpublished data) along with significant levels of fruit abortion, probably a result of inbreeding depression.
Small-fragment harbors an amount of genetic diversity similar to the continuous and larger fragment, as shown by alpha diversity estimates. Despite allele frequencies among patches overlapped in an average level of around 80 % (\(\bar{\omega }\) = 0.812), the small divergence in allele composition among patches was significant, showing that the small-fragment, even being small and degraded, is a valuable target for in situ conservation as it maintains levels of genetic diversity similar to larger populations. The contribution of small remnants or even isolated trees in conserving genetic diversity in fragmented landscapes was also observed in other tree species (Finger et al. 2012; Ganzhorn et al. 2015; Ribeiro et al. 2005; White et al. 2002). Furthermore, our results highlight that seed trees in small and degraded fragments can be relevant sources of genetic diversity for seed collection aimed at restoration initiatives, especially in severely fragmented landscapes (the BAF reality in our region) where large continuous forests no longer exist. Choosing seed trees scattered in small but well sprawling fragments to collect seeds for local restoration plans could be a valuable strategy to at least partially recover the levels of genetic diversity of the original landscape. The pairwise genetic divergence (δ) among adults varied across comparisons (Fig. 4) and confirmed the spatial genetic structure revealed in sPCA (Fig. 2). Two distinct groups were clearly identified in the small-fragment; the São Roque farm (section B) is distinct from the three sections (C, D, and E) in the campus area. If we consider C. langsdorffii as an indicator species, this fine-scale overview of genetic diversity distribution provided by both sPCA and alpha–beta-gamma statistics can be used to guide a specific in situ conservation strategy for this area. To maintain the overall levels of genetic diversity, at least three patches should be preserved, the large-fragment (section A) and two patches in small-fragment: São Roque farm (section B) and one of the sections in campus area (C, D or E). The choice should also consider habitat configuration (spatial distribution of remnants in the matrix), in order to improve functional connectivity and increase population persistence (Villard et al. 2014).
We observed a decrease in genetic diversity in seedlings in comparison to adults (Fig. 3; Table 3). Although adult populations are expected to show higher levels of genetic diversity due to overlap of generations, our results clearly show that the gene pool that formed adult populations was greater than the gene pool represented in seedlings (gamma diversity was almost 30 % greater in adults than in seedlings). This observation is in line with theoretical expectations of habitat fragmentation (Young et al. 1996) and was also observed in other trees studied in fragmented forests (Dayanandan et al. 1999; Finger et al. 2012; Isagi et al. 2007; Sebbenn et al. 2011). We also detected a smaller degree of genetic divergence among patches in seedlings than in adults, showing that the studied fragments are somehow connected by gene flow, but they may not be receiving propagules from farther sources. Pollination and seed dispersal vectors are commonly observed in our landscape, able to forage at distances encompassed in this study (~2 km), and capable of crossing the agricultural matrix between these fragments. Perhaps, the high abundance of local adults and the synchronic flowering (Pedroni et al. 2002) limit effective pollen flow from farther fragments, as local trees available as pollen sources are abundant (Sork and Smouse 2006) and generalist pollinating bees tend to forage narrowly when local pollen availability is large. This hypothesis could also explain the decrease in genetic diversity in seedlings compared to adults. At a broader scale, current levels of gene flow have not been sufficient to prevent the loss of genetic diversity across generations. Part of the loss of genetic diversity in smaller life stages such as seedlings may be due to increasing regeneration near adults, which can also help to explain the high local abundance of C. langsdorffii in the sampled fragments.
In our landscape, C. langsdorffii does not seem to be affected by negative distance/density dependent effects (based on distance from nearest adult plants) as it is in another BAF region (Oliveira-Filho et al. 1996). Certain negative density-dependent mortality factors such as seed predation, seedling herbivory and pathogenic disease spread can be relaxed in disturbed tropical forests (Dirzo and Miranda 1991; Terborgh 2013), enabling plant regeneration beneath parental plants to occur more frequently than in old-growth forests (e.g. Harms et al. 2000). Forest disturbances such as habitat loss, fragmentation and hunting have pervasive effects on seed predators and dispersers (Dirzo et al. 2014; Peres and Palacios 2007) which can trigger a decrease in the amount of seeds dispersed and the distances achieved by those seeds, influencing plant regeneration (Cordeiro and Howe 2003; Markl et al. 2012). Seedlings of C. langsdorffii seem to be largely tolerant to the harsh environmental conditions found at fragment edges, as demonstrated by the high regeneration rate (1.31 %) in these small fragments and high seedling survivor probability (>80 %). Thus, the tolerance of C. langsdorffii to high densities and edge conditions would make this plant less sensitive to BAF fragmentation and disturbance. It may also help explain the large number of adults found per hectare in the sampled fragments (see also Andreazzi et al. 2012). Although these results hint that this C . langsdorffii population is somewhat resilient to habitat fragmentation and that these small and degraded fragments are valuable targets for in situ conservation, the maintenance of genetic diversity in the long term is compromised by the lack of connectivity on a broader scale. Restoration plans, even at a small scale, are necessary to promote fragment connectivity and spatially expand opportunities for the fairly restricted gene flow observed in this severely fragmented BAF region.
The integration of regeneration ecology and molecular genetics methodologies provided us a deeper overview of the effects of habitat fragmentation in C. langsdorffii than using only one approach. It allowed us to conclude that solid recruitment is not synonymous of long-term population persistence, as the maintenance of genetic diversity over time can be compromised even when the species is fairly common and is regenerating. Furthermore, we proved that alpha–beta-gamma statistics are a valuable tool to address the conservation status of fragmented populations. We recommend its application in future studies on conservation genetics and habitat fragmentation.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188. doi:10.1111/j.1365-294X.2008.03971.x
Andreazzi CS, Pimenta CS, Pires AS, Fernandez FAZ, Oliveira-Santos LG, Menezes JFS (2012) Increased productivity and reduced seed predation favor a large-seeded palm in small Atlantic Forest fragments. Biotropica 44:237–245
Bacles CF, Jump AS (2011) Taking a tree’s perspective on forest fragmentation genetics. Trends Plant Sci 16:13–18. doi:10.1016/j.tplants.2010.10.002
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol Notes 5:453–455
Christianini AV, Oliveira PS (2013) Edge effects decrease ant-derived benefits to seedlings in a neotropical savanna. Arthropod-Plant Interactions 7:191–199. doi:10.1007/s11829-012-9229-9
Ciampi AY, Brondani RVP, Grattapaglia D (2000) Desenvolvimento de marcadores microssatélites para Copaifera langsdorffii Desf. (copaíba) Leguminosae—Caesalpinoideae e otimização de sistemas fluorescentes de genotipagem multiloco. vol 16. Embrapa Recursos Genéticos e Biotecnologia, Brasília
Cordeiro NJ, Howe HF (2003) Forest fragmentation severs mutualism between seed dispersers and an endemic African tree. Proc Natl Acad Sci USA 100:14052–14056. doi:10.1073/pnas.2331023100
Corrêa LS, Cardoso-Leite E, Castello ACD, Coelho S, Kortz AR, Villela FNJ, Koch I (2014) Estrutura, composição florística e caracterização sucessional em remanescente de floresta estacional semidecidual no sudeste do. Brasil Revista Árvore 38:799–809
Costa JAS (2007) Estudos taxonômicos, biossistemáticos e filogenéticos em Copaifera L. (Leguminosae: Detarieae) com ênfase nas espécies do brasil extra-amazônico. Universidade Estadual de Feira de Santana
Dabrowski MJ, Pilot M, Kruczyk M, Zmihorski M, Umer HM, Gliwicz J (2014) Reliability assessment of null allele detection: inconsistencies between and within different methods. Mole Ecol Resour 14:361–373. doi:10.1111/1755-0998.12177
Dabrowski MJ, Bornelov S, Kruczyk M, Baltzer N, Komorowski J (2015) ‘True’ null allele detection in microsatellite loci: a comparison of methods, assessment of difficulties and survey of possible improvements. Mole Ecol Resour 15:477–488. doi:10.1111/1755-0998.12326
Dayanandan S, Dole J, Bawa K, Kesseli R (1999) Population structure delineated with microsatellite markers in fragmented populations of a tropical tree, Carapa guianensis (Meliaceae). Mol Ecol 8:1585–1592
de Mello K, Petri L, Cardoso-Leite E, Toppa RH (2014) Cenários ambientais para o ordenamento territorial de áreas de preservação permanente no município de Sorocaba, SP. Revista Árvore 38:309–317
Demšar U, Harris P, Brunsdon C, Fotheringham AS, McLoone S (2013) Principal component analysis on spatial data: an overview. Ann Assoc Am Geogr 103:106–128. doi:10.1080/00045608.2012.689236
Dirzo R, Miranda A (1991) Altered patterns of herbivory and diversity in the forest understory: A case study of the possible consequences of contemporary defaunation. In: Price PW, Lewinsohn TM, Fernandes GW, Benson WW (eds) Plant-animal interactions: Evolutionary ecology in tropical and temperate regions. Wiley, New York, pp 273–287
Dirzo R, Young HS, Galetti M, Ceballos G, Isaac NJB, Coll B (2014) Defaunation in the Anthropocene. Science 345:401–406
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Ewers RM, Thorpe S, Didham RK (2007) Synergistic interactions between edge and area effects in a heavily fragmented landscape. Ecology 88:96–106
Finger A, Kettle CJ, Kaiser-Bunbury CN, Valentin T, Mougal J, Ghazoul J (2012) Forest fragmentation genetics in a formerly widespread island endemic tree: vateriopsis seychellarum (Dipterocarpaceae). Mol Ecol 21:2369–2382. doi:10.1111/j.1365-294X.2012.05543.x
Fletcher RJ Jr (2005) Multiple edge effects and their implications in fragmented landscapes. J Anim Ecol 74:342–352. doi:10.1111/j.1365-2656.2005.00930
Freitas CV, Oliveira PE (2002) Biologia reprodutiva de Copaifera langsdorffii Desf. (Leguminosae, Caesalpinioideae). Revista Brasileira de Botânica 25:311–321
Ganzhorn SM, Perez-Sweeney B, Thomas WW, Gaiotto FA, Lewis JD (2015) Effects of fragmentation on density and population genetics of a threatened tree species in a biodiversity hotspot. Endanger Species Res 26:189–199. doi:10.3354/esr00645
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Guillot G, Estoup A, Mortier F, Cosson JF (2005a) A spatial statistical model for landscape genetics. Genetics 170:1261–1280. doi:10.1534/genetics.104.033803
Guillot G, Mortier F, Estoup A (2005b) Geneland: a computer package for landscape genetics. Mol Ecol Notes 5:712–715. doi:10.1111/j.1471-8286.2005.01031.x
Haddad NK, Brudvig LA, Clobert J, Davies KF, Gonzalez A et al (2015) Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci Adv. doi:10.1126/sciadv.1500052
Hamrick JL (2004) Response of forest trees to global environmental changes. For Ecol Manage 197:323–335. doi:10.1016/j.foreco.2004.05.023
Harms KE, Wright SJ, Calderón O, Hernández A, Herre EA (2000) Pervasive density-dependent recruitment enhances seedling diversity in a tropical forest. Nature 404:493–495
Isagi Y, Tateno R, Matsuki Y, Hirao A, Watanabe S, Shibata M (2007) Genetic and reproductive consequences of forest fragmentation for populations of Magnolia obovata. Ecol Res 22:382–389. doi:10.1007/s11284-007-0360-5
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. doi:10.1093/bioinformatics/btn129
Jombart T, Devillard S, Dufour AB, Pontier D (2008) Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity (Edinb) 101:92–103. doi:10.1038/hdy.2008.34
Kortz AR, Coelho S, Castello ACD, Corrêa LS, Cardoso-Leite E, Koch I (2014) Wood vegetation in Atlantic rain forest remnants in Sorocaba (São Paulo, Brazil). Check List 10:344–354
Kramer AT, Ison JL, Ashley MV, Howe HF (2008) The paradox of forest fragmentation genetics. Conserv Biol 22:878–885. doi:10.1111/j.1523-1739.2008.00944.x
Laurance WF (2002) Hyperdynamism in fragmented habitats. J Veg Sci 13:595–602
Laurance WF et al (2011) The fate of Amazonian forest fragments: a 32-year investigation. Biol Conserv 144:56–67. doi:10.1016/j.biocon.2010.09.021
Lowe AJ, Boshier D, Ward M, Bacles CF, Navarro C (2005) Genetic resource impacts of habitat loss and degradation; reconciling empirical evidence and predicted theory for neotropical trees. Heredity 95:255–273. doi:10.1038/sj.hdy.6800725
Malcolm JR (1994) Edge effects in Central Amazonian forest fragments. Ecology 75:2438–2445
Manoel RO, Alves PF, Dourado CL, Gaino APSC, Freitas MLM, Moraes MLT, Sebbenn AM (2012) Contemporary pollen flow, mating patterns and effective population size inferred from paternity analysis in a small fragmented population of the Neotropical tree Copaifera langsdorffii Desf. (Leguminosae-Caesalpinioideae). Conserv Genet 13:613–623. doi:10.1007/s10592-011-0311-0
Markl JS et al (2012) Meta-analysis of the effects of human disturbance on seed dispersal by animals. Conserv Biol 26:1072–1081. doi:10.1111/j.1523-1739.2012.01927.x
Martins K, Santos JD, Gaiotto FA, Moreno MA, Kageyama PY (2008) Estrutura genética populacional de Copaifera langsdorffii Desf. (Leguminosae - Caesalpinioideae) em fragmentos florestais no Pontal do Paranapanema, SP, Brasil. Revista Brasileira de Botânica 31:61–69. doi:10.1590/S0100-84042008000100007
Motta Junior JC, Lombardi JA (1990) Aves como agentes dispersores de copaíba (Copaifera langsdorffii, Caesalpiniaceae) em São Carlos, estado de São Paulo. Ararajuba 1:105–106
Murcia C (1995) Edge effects in fragmented forests: implications for conservation. Trends Ecol Evol 10:58–62. doi:10.1016/S0169-5347(00)88977-6
Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858. doi:10.1038/35002501
Oliveira-Filho A, Camisão-Neto AA, Volpato MML (1996) Structure and dispersion of four tree populations in an area of montane semidecidous forest in southeastern Brazil. Biotropica 28:762–769
Pedroni F, Sanchez M, Santos FAM (2002) Fenologia da copaíba (Copaifera langsdorffii Desf.—Leguminosae, Caesalpinioideae) em uma floresta semidecídua no sudeste do Brasil. Revista Brasileira de Botânica 25:183–194
Peres CA, Palacios E (2007) Basin-wide effects of game harvest on vertebrate population densities in amazonian forests: implications for animal-mediated seed dispersal. Biotropica 39:304–315
Pütz S, Groeneveld J, Alves LF, Metzger JP, Huth A (2011) Fragmentation drives tropical forest fragments to early successional states: a modelling study for Brazilian Atlantic forests. Ecol Model 222:1986–1997. doi:10.1016/j.ecolmodel.2011.03.038
R CoreTeam (2015) R: A language and environment for statistical computing. Vienna, Austria
Ribeiro RA, Simoes Ramos AC, De Lemos Filho JP, Lovato MB (2005) Genetic variation in remnant populations of Dalbergia nigra (Papilionoideae), an endangered tree from the Brazilian Atlantic Forest. Ann Bot 95:1171–1177. doi:10.1093/aob/mci128
Ribeiro MC, Metzger JP, Martensen AC, Ponzoni FJ, Hirota MM (2009) The Brazilian Atlantic Forest: how much is left, and how is the remaining forest distributed? Implications for conservation. Biol Conserv 142:1141–1153. doi:10.1016/j.biocon.2009.02.021
Santos BA, Peres CA, Oliveira MA, Grillo A, Alves-Costa CP, Tabarelli M (2008) Drastic erosion in functional attributes of tree assemblages in Atlantic forest fragments of northeastern Brazil. Biol Conserv 141:249–260. doi:10.1016/j.biocon.2007.09.018
Saunders DA, Hobbs RJ, Margules CR (1991) Biological consequences of ecosystem fragmentation: a review. Conserv Biol 5:18–32
Scofield DG, Smouse PE, Karubian J, Sork VL (2012) Use of alpha, beta, and gamma diversity measures to characterize seed dispersal by animals. Am Nat 180:719–732. doi:10.1086/668202
Sebbenn AM et al (2011) Low levels of realized seed and pollen gene flow and strong spatial genetic structure in a small, isolated and fragmented population of the tropical tree Copaifera langsdorffii. Desf Hered (Edinb) 106:134–145. doi:10.1038/hdy.2010.33
Sheil D, Burslem DFRP, Alder D (1995) The interpretation and misinterpretation of mortality rate measures. J Ecol 83:331–333
Sork VL, Smouse PE (2006) Genetic analysis of landscape connectivity in tree populations. Landsc Ecol 21:821–836. doi:10.1007/s10980-005-5415-9
Sork VL, Smouse PE, Grivet D, Scofield DG (2015) Impact of asymmetric male and female gamete dispersal on allelic diversity and spatial genetic structure in valley oak (Quercus lobata Née). Evol Ecol 29(6):927–945
Tabarelli M, Lopes AV (2008) Edge-effects drive tropical forest fragments towards an early-successional system. Biotropica 40:657–661
Tabarelli M, Aguiar AV, Ribeiro MC, Metzger JP, Peres CA (2010) Prospects for biodiversity conservation in the Atlantic Forest: lessons from aging human-modified landscapes. Biol Conserv 143:2328–2340. doi:10.1016/j.biocon.2010.02.005
Tarazi R, Sebbenn AM, Kageyama PY, Vencovsky R (2013a) Edge effects enhance selfing and seed harvesting efforts in the insect-pollinated Neotropical tree Copaifera langsdorffii (Fabaceae). Heredity 110:578–585. doi:10.1038/hdy.2013.8
Tarazi R, Sebbenn AM, Kageyama PY, Vencovsky R (2013b) Long-distance dispersal in a fire- and livestock-protected savanna. Ecol Evol 3:1003–1015. doi:10.1002/ece3.515
Templeton AR, Shaw K, Routman R, Davis SK (1990) The genetic consequences of habitat fragmentation. Ann Mo Bot Gard 77:13–27
Terborgh J (2013) Using Janzen-Connell to predict the consequences of defaunation and other disturbances of tropical forests. Biol Conserv 163:7–12. doi:10.1016/j.biocon.2013.01.015
Turner IM (1996) Species loss in fragments of tropical rain forest: a review of the evidence. J Appl Ecol 33:200–209
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. doi:10.1111/j.1471-8286.2004.00684.x
Villard M-A, Metzger JP, Saura S (2014) REVIEW: beyond the fragmentation debate: a conceptual model to predict when habitat configuration really matters. J Appl Ecol 51:309–318. doi:10.1111/1365-2664.12190
White GM, Boshier DH, Powell W (2002) Increased pollen flow counteracts fragmentation in a tropical dry forest: an example from Swietenia humilis Zuccarini. Proc Natl Acad Sci USA 99:2038–2042. doi:10.1073/pnas.042649999
Whittaker RH (1972) Evolution and measurement of species diversity. Taxon 21:213–251
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:6
Acknowledgments
We thank Ivonir Piotrowski, João Paulo de S. Curti and Thiago P. Telatin for helping in field work, and the owners of Fazenda São Roque, Fazenda Flores, and Ecoresidencial Fazenda Jequitibá for authorizing field work in their farms. We also thank Victoria Sork for her insightful comments on the first versions of this manuscript and Carla Américo for drawing the map. Two anonymous reviewers helped improve the manuscript. We finally thank São Paulo Research Foundation (FAPESP) grant 2012/01274-4 and FAPESP-University of Florida grant 2013/50119-4 for financial support. AFF received scientific initiation scholarships from National Council for Scientific and Technological Development (CNPq) in 2012 and from FAPESP in 2013 (Grant 2012/20363-8).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Martins, K., Kimura, R.K., Francisconi, A.F. et al. The role of very small fragments in conserving genetic diversity of a common tree in a hyper fragmented Brazilian Atlantic forest landscape. Conserv Genet 17, 509–520 (2016). https://doi.org/10.1007/s10592-015-0800-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-015-0800-7