
Abstract
Understanding the extent to which diversity at immunologically important genes is reduced by demographic perturbations such as population bottlenecks, and the resulting consequences this has on individual fitness, is of fundamental importance for the effective management of genetic resources in natural populations. Toll-like receptors are key immunological genes with well-established links to fitness. Here, levels of allelic diversity and heterozygosity at the toll-like receptor 4 locus (Tlr4) were characterised across 280 water voles (Arvicola amphibius) from an isolated, island population in north west Scotland that went through a severe population bottleneck between 2004 and 2006 that eroded neutral microsatellite variation. Two functional Tlr4 alleles were resolved prior to the population crash at frequencies close to parity and an excess of heterozygote genotypes relative to Hardy–Weinberg expectations. Through the population bottleneck both alleles were retained with genotype frequencies conforming to Hardy–Weinberg expectations. Tlr4 genotype was significantly associated with gamasid mite, flea (Megabothris walkeri) and sheep tick larva (Ixodes ricinus) burdens among individuals, suggesting a mechanism through which parasite mediated selection could affect Tlr4 diversity. The results are examined with recourse to the extent that they are consistent with the effects of genetic drift and balancing selection, and their significance is discussed in relation to identifying target genes that assay ecological and adaptively meaningful genetic variation in a conservation context.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Population genetics theory highlights that small populations are at increased risk of experiencing loss of genetic diversity through the effects of random genetic drift (Wright 1931; Nei et al. 1975; Allendorf 1986). The knock-on effects of this process can cause a reduction in individual fitness and population viability through the negative effects of inbreeding depression, mediated though reduced heterozygosity and the fixation of deleterious recessive mutations (Charlesworth and Charlesworth 1987; Piertney and Webster 2008). Moreover, low levels of genetic diversity may compromise a population’s evolutionary potential, and thus the ability to adapt to a changing environment and associated selection pressures (Ridley 2003).
Demographic perturbations such as population bottlenecks and founder events are expected to erode genetic variation given the extent to which they can reduce population size (Wright 1931; Nei et al. 1975; Kimura 1983; Dlugosch and Parker 2008). Several studies have demonstrated the expected loss of both allelic diversity and heterozygosity that is associated with population bottlenecks for neutral molecular markers (Keller et al. 2001; Wisely et al. 2002; Taylor et al. 2012). However, assays of neutral genetic diversity in population surveys are not always the most reliable predictors of adaptive genetic variation (Merila and Crnokrak 2001; Reed and Frankham 2001; Piertney and Webster 2008; Alcaide and Edwards 2011; Bollmer et al. 2011) so it is unclear whether the same effects are observed for adaptive polymorphisms. In some instances it has been demonstrated that even in small populations, selection can act in opposition with genetic drift to preserve variation at adaptive loci (Oliver and Piertney 2012). As such, directly assaying levels of diversity at key genes that explicitly influence fitness can reveal adaptive variation driven by selection that may not be observed at neutral molecular markers (Allendorf et al. 2010; Ouborg et al. 2010).
A common target for examining adaptively important and ecologically relevant genetic variation in natural populations has been the Major Histocompatibility Complex (MHC), as it is often observed to display a high level of diversity and is associated with fitness-related traits such as parasite burden and sexual selection (Bernatchez and Landry 2003; Sommer 2005; Piertney and Oliver 2006; Consuegra and Garcia de Leaniz 2008; Oliver et al. 2009a, b; Dunn et al. 2013). However, the MHC represents only a small portion of the immunogenome as well as the broader adaptive fraction of the overall genome. The advent of post-genomics technologies and analyses has greatly enhanced the potential to identify and target key genome regions outside the MHC that represent functional genetic variation of ecological relevance (Blackwell 2001; Piertney and Webster 2008; Horner et al. 2010). This is an essential step forward, given that there are many other non-MHC immune function genes that play pivotal roles in the vertebrate immune system (Acevedo-Whitehouse and Cunningham 2006).
An important class of non-MHC immune function genes are the toll-like receptors (TLRs). TLRs are an essential family of type 1 transmembrane glycoproteins that act as pattern recognition receptors (PRRs) and play an important role in the innate immune response (Akira et al. 2001; Brownlie and Allan 2011; Grueber et al. 2013; Fornusková et al. 2013). They are involved in the initial recognition of invading infectious agents (Tschirren et al. 2013) and act as a host’s front-line defence against a wide range of pathogens (Uematsu and Akira 2008; Iwasaki and Medzhitov 2010; Alcaide and Edwards 2011). They also act as an important bridge between the processes involved in innate and acquired immunity (Akira et al. 2001; Bollmer et al. 2011). TLRs are typically composed of a pathogen-recognition extracellular domain, a short transmembrane segment and an intracellular toll/interleukin-1 signalling domain (Gay and Gangloff 2007; Werling et al. 2009; Alcaide and Edwards 2011). The extracellular signalling domain is composed of a leucine-rich repeat motif which plays a vital role in detecting the distinctive features of invading microbes, so called “pathogen associated molecular patterns” (Takeuchi et al. 1999; Werling et al. 2009; Alcaide and Edwards 2011; Tschirren et al. 2013). Once bound to an invading pathogen, TLRs orchestrate the first volley of immune defence by initiating a signalling cascade which is mediated by specific adaptor proteins (Akira et al. 2001; Alcaide and Edwards 2011). The resulting signal cascade causes the activation of transcription factors and the production of cytokines that ultimately result in the inflammatory response within the host (Kawai and Akira 2005; Bollmer et al. 2011; Tschirren et al. 2011), and the activation of the antigen-specific adaptive immune response (Iwasaki and Medzhitov 2010).
There are 10–13 different TLRs within most mammal species, which are specific to a range of different pathogens (Roach et al. 2005; Tschirren et al. 2013). Several of these TLR genes are thought to evolve under the effects of balancing selection (Ferrer-Admetlla et al. 2008) and show high levels of allelic variation (Alcaide and Edwards 2011; Bergman et al. 2012; Grueber et al. 2012). One of the best characterised TLRs is toll-like receptor 4 (Tlr4), which recognises lipopolysaccharides in the outer membrane of gram negative bacteria (Akira et al. 2001; Shi et al. 2006; Werling et al. 2009). As Tlr4 is known to play an important role in a host’s defence against parasites, genetic polymorphism at this locus has been recognised as a potential candidate for investigating the signatures of selection acting upon innate immune defence genes in natural populations (Hughes and Friedman 2008; Tschirren et al. 2013).
In this study, we investigate Tlr4 diversity in an isolated population of water voles (Arvicola amphibius) located on the small island of Coiresa in North West Scotland, UK. Specifically we examine the extent to which diversity is affected by drift mediated through long-term isolation of the population and through the effects of a population bottleneck that spanned 3 years which, at its lowest point, reduced the population size down to just six individuals (five of which were adults and of those there were three males and two pregnant females). The population collapse and subsequent recovery coincided with the presence of sheep (Ovis aries) on the island that were introduced in the autumn of 2004, and removed in the summer of 2005. The sheep had a substantial impact on the island’s grass coverage which severely reduced the food supply and amount of suitable habitat coverage for the water voles, causing the population to crash. It has previously been shown that levels of diversity at six microsatellite markers were reduced within this population by the bottleneck, consistent with the effects of random genetic drift in a small isolated population (Oliver and Piertney 2012). Prior to the population bottleneck, microsatellite allelic variation was already low relative to the levels of variation present at the same loci in a large mainland population (Stewart et al. 1998; Oliver and Piertney 2012). During the bottleneck further variation was lost and there was no significant deviations from neutral expectations before, during or after the population bottleneck (Oliver and Piertney 2012). These changes were in contrast to patterns observed at a class II MHC locus (Oliver et al. 2009a, b) where it was shown that balancing selection countered the effects of drift to retain variation through the bottleneck. This was achieved by heterozygote individuals being significantly fitter than their homozygote counterparts (Oliver et al. 2009a; Oliver and Piertney 2012). A previous study highlighted that host-parasite interaction were likely drivers of balancing selection, with different alleles at the MHC DQA locus explaining variance in ectoparasite burdens, with MHC heterozygote voles having the lowest overall parasite load in mixed infections (Oliver et al. 2009a).
We examine the dynamics of Tlr4 through the population bottleneck to test whether it displays patterns more similar to neutral microsatellites or MHC variation. We examine both allelic variation and heterozygosity prior to the population crash, and for the 3 years of the crash and the time required for the population to recover back to pre-bottleneck density. Data simulation was conducted to estimate heterozygosity and allelic frequency assuming only random genetic drift was operating. By using these simulations assessment can be made as to whether Tlr4 gene dynamics matches neutral expectations.
We also examine the extent to which Tlr4 genotype explains individual variation in flea (Megabothris walkeri and Ctenopthalmus nobilis), tick (Ixodes ricinus larvae and nymphs) and mite (Gamasidae) parasite burdens, and prevalence of the bacterial microparasite Bartonellla. Tlr4 genotype has previously been shown to influence levels of parasite burden (Garantziotis et al. 2008; Tschirren et al. 2011; Bollmer et al. 2011; Tschirren et al. 2013).
We predict that: (1) Tlr4 variation will show patterns of diversity inconsistent with neutral expectations; and (2) Tlr4 genotype will significantly explain variation in the number of parasites among individuals.
Materials and methods
Study site and sample collection
The focal water vole population is on the small (~2000 m2) island of Coiresa (56°08′ N, 5°37′ W), in the Sound of Jura on the west coast of Scotland. Intensive, long-term sampling of the island since 2003 has confirmed a lack of immigration into the population, with levels of microsatellite-derived genetic differentiation with neighbouring islands suggesting isolation for much longer periods (Telfer et al. 2003). The vast majority of voles in this population do not survive to a second breeding season so generations are effectively discrete across years. Sampling occurred during each Spring and Autumn between May 2004 and October 2006 as described in Oliver et al. 2009a, b. In brief, a targeted trapping grid was established, consisting of four transects of 25–30 baited Elliot live traps that were positioned close to the signs of water vole activity (clippings, latrines, runs, and burrows). These were checked every 24 h over a three to four night period. For all individuals caught, sex, reproductive status and weight (g) was recorded. Voles that were classed as reproductively inactive were assigned as juveniles while voles that were reproductively active were classed as adults. Tissue sampling for DNA extraction and the quantification of parasite burdens (i.e. direct counts) of mites (Gamasidae), fleas (M. walkeri and C. nobilis) tick (I. ricinus) nymphs and larvae were as detailed in Oliver et al. 2009a, b. Blood samples were screened for microparasites of the bacterial genus Bartonella and these results were recorded as prevalence data (Oliver et al. 2009a, b).
Molecular analysis
Genomic DNA was extracted using the salting out procedure described in Mullenbach et al. (1989) with dilution to 10–20 ng µl−1 in 10 mM Tris–HCl (pH 7.0). The polymerase chain reaction (PCR) was used to amplify a 1000 bp region of the extracellular domain that encodes pathogen recognition using the primers (5′-3′) F 5′-AAA TGG CTG GCA ATT CTT TC -3′ and R 5′-AGT CKT CTC CAG AAG ATG TGC-3′ (Turner et al. 2012). As this region is directly responsible for interacting with pathogen-derived structures it is predicted to be the most polymorphic (Fornusková et al. 2013). PCRs were carried out in an MJ Research PTC-100 thermocycler in a total volume of 20 µl consisting of 25 ng template DNA, 2.5 mM MgCl2, 16 mM (NH4)2SO4, 67 mM Tris–HCl, 0.2 mM of each nucleotide, 0.5 µM of each primer and 0.5 U of Taq DNA polymerase (Sigma-Aldrich). The PCR conditions were: an initial denaturation step at 95 °C for 5 min followed by 30 cycles of annealing at 65.3 °C for 30 s, elongation at 72 °C for 30 s and denaturation at 95 °C for 30 s. An extra 5 min elongation step at 72 °C followed the final cycle. PCR success was confirmed on 2 % 1 × TBE agarose gels stained with WebGreen DNA stain (Web Scientific). Amplified products were run alongside a Hyperladder IV size standard (Bioline) and compared with negative controls to check for contamination.
Samples were genotyped using denaturant gradient gel electrophoresis (DGGE). For each individual, 5 µl of PCR product was mixed with 5 µl 2.5× loading dye (Bioline) and loaded onto a 16 × 20 cm, 1 mm thick 8 % polyacrylamide gel containing a 20–70 % gradient of denaturing agents (urea and formamide) and run in a Dcode™ universal mutation detection system (Bio-Rad Laboratories, Hercules, CA) in 6.5 L of 1 × TAE buffer at 50 V for 14 h at 60 °C. Products were visualised using a standard silver staining protocol (Bassam and Gresshoff 2007). DGGE banding patterns were scored as 1/1, 1/2, 2/2 to represent the three possible genotypes. For each banding pattern phenotype that was resolved, products from five individuals were purified using the QIAquick PCR purification kit (Qiagen Ltd) according to the manufacturer’s instructions and sequenced on an automated DNA sequencer by Eurofins MWG, London, using the forward PCR primer. To resolve allele sequences from heterozygote genotypes when multiple variable sites were resolved, PCR amplicons were cloned using the pGEM®-T Easy Vector system following manufacturer’s instructions (Promega), and then DNA sequenced using M13 primers.
One DGGE banding phenotype failed to resolve some of the underlying DNA sequence variation. A DNA sequence variant fell within an MboII restriction enzyme recognition site and so this enzyme was used in a restriction fragment length polymorphism (RFLP) assay to assign ambiguous DGGE patterns to genotype. The PCR amplicon was digested overnight at 37 °C with 1 unit of MboII, and the product run out on a WebGreen stained 2 % agarose gel alongside a Hyperladder IV standard (Bioline).
Statistical analysis
Two separate approaches were used to examine departures of genetic variation from neutral expectations. First, the inbreeding coefficient F IS was estimated using FSTAT v 2.9.3.2 (Goudet 2005) to detect deviations from Hardy–Weinberg equilibrium (HWE), the significance of which was tested by randomised permutation. Second, the Ewens–Watterson (E–W) test of neutral sequence evolution (Watterson 1978) based on sequence haplotypes was used as implemented in Arlequin v 3.5 (Excoffier et al. 2005). Linkage disequilibrium between the Tlr4 locus and the previously examined MHC locus (Oliver et al. 2009a, b) was tested at each sampling event using Genepop v 4.2 (Rousset 2008) using the log likelihood ratio statistic and the default Markov chain parameters (dememorization number = 1000, number of batches = 100, number of iterations per batch = 1000). To test for evidence of a long-term signature of selection the rates of synonymous and non-synonymous mutations were calculated with a Jukes Cantor correction. To calculate nucleotide diversity fragments were aligned and trimmed to ~750 bp. A Z-test was used to examine whether the frequency of non-synonymous mutations per non-synonymous site was significantly greater than synonymous mutations per synonymous site. Calculations were undertaken in DNAsp v 5 (Librado and Rozas 2009).
The evolutionary relationships between the A. amphibius alleles with homologous sequences from closely related species was resolved with a maximum likelihood phylogenetic tree using MEGA6 (Tamura et al. 2013). The optimised model of sequence evolution was ascertained using ModelTest, and bootstrapping (10,000 iterations) used to assess the stability of internal nodes.
Simulation studies
Simulations were undertaken to provide estimates of heterozygosity and allele frequency for neutral polymorphisms prior to, and through the course of the bottleneck. By comparing the simulated data to the observed data at the Tlr4 locus assessment can be made as to whether Tlr4 gene dynamics matches neutral expectations. For both the pre-bottleneck and bottleneck simulations a population level simulation and an individual-based simulation were created. At the population level only the initial population allele frequency was specified and at the end of the simulation the difference between the final allele frequencies was calculated. The rationale behind this approach was that genetic drift would lead to large differences in allele frequency as one allele is forced towards fixation, whereas balancing selection would aim to maintain parity in allele frequency when retaining both alleles. In the individual-based simulations genotypes were assigned to each individual so the difference in allele frequency and the proportion of heterozygotes could be calculated. A Chi squared test was used to establish if the population conformed to Hardy–Weinberg expectations. All simulations were created within the R statistical environment v 3.1.2 (R Core team 2013).
Pre-bottleneck simulations
The first simulation generated allele frequencies for a two allele locus in a population maintained at 100 individuals, with a starting allele frequency of p = q = 0.5, equal sex ratios (s.r. = 0.5) and random mating over 100 generations. The simulation was run 10,000 times and the difference between the two allele frequencies was calculated and compared to the observed May 2004 pre-bottleneck Tlr4 allele difference of 0.04.
The second simulation used an individual-based approach. A starting population of 100 individuals with an even allele frequency (p = q = 0.5), even sex ratio and a heterozygosity proportion of 0.5 (H = 0.5) was established. This was used to seed a second generation assuming a year-on-year survival rate of 0.1, a constant population size of n = 100 and random mating among individuals. Alleles were sampled at random from parental genotypes and apportioned according to Mendelian expectation among the offspring. This process was repeated for 100 generations, and repeated 1000 times. The difference in final allele frequencies and the proportion of heterozygotes were recorded for each run. To test if the final population was within Hardy–Weinberg equilibrium a Chi square test was performed on each of the final populations. Populations with a p value < 0.05 were deemed to be out-with Hardy–Weinberg expectations.
Simulating the bottleneck
The third simulation was used to generate allele frequencies throughout the course of the population bottleneck. An initial population (generation 0) was generated that had the starting population size and allele frequency observed in May 2004 (n = 81, p = 0.52). For each of the subsequent generations the population sizes mirrored those observed during the bottleneck (generation 1 n = 6; generation 2 n = 14; generation 3 n = 70). For the final generation the difference in allele frequency was calculated and compared to the observed October 2006 Tlr4 value of 0.32. This simulation was repeated 10,000 times.
The final simulation utilised the individual-based approach to generate the proportion of heterozygous as well as the allele frequencies throughout the population bottleneck. An initial population (generation 0) was generated using the parameters observed for the May 2004 population (n = 81, p = 0.52, H = 0.8, s.r. = 0.5). Once the initial population had been generated the parameters for the subsequent generations were specified (generation 1 n = 6, survivors = 0, s.r. = 0.5; generation 2 n = 14, survivors = 2, survivor s.r. = 0.5, s.r. = 0.43; generation 3 n = 70, survivors = 4, survivor s.r. = 0.36, s.r. = 0.54). Again, offspring genotypes were generated by randomly sampling parental genotypes with sampling probabilities based on Mendelian expectations. The simulation was run 10,000 times and the difference in final allele frequencies, the proportion of heterozygotes and Chi square p values were recorded for the last generation of each run.
Statistical models
Statistical models were constructed to examine the differences between Tlr4 genotypes in parasite burden of different parasite species (M. walkeri and C. nobilis fleas, I. ricinus larvae, I. ricinus nymphs, microparasites of the bacterial genus Bartonella and mites of the genus Gamasidae) while controlling for the potentially confounding influence of sex, reproductive status, month and year. Collinearity between explanatory variables was identified between vole weight and reproductive status, and vole weight and sex. The potentially confounding effect of these variables was removed during model selection by ensuring that collinear explanatory variables were not used in the same model.
In all instances, the error structure of the parasite response variable was not assumed to be normally distributed as they were either count or presence-absence variables. As such, models were fitted using either a Poisson error structure (for count data) or a binomial error structure (for presence/absence) data. The dataset included repeat measurements from some individuals that had survived between sampling periods. In order to account for this non-independence, generalised linear mixed models (GLMM) were used where an indicator variable of individual ID was included as an additive random effect in addition to the fixed effects. All models were fitted with the function glmer using the package lme4 v 1.1-7 (Bates et al. 2014) within the R statistical environment v 3.1.2 (R Core team 2013).
Initial models for each response variable were constructed that included the main effects of Tlr4 genotype, sex, reproductive status, month and year. In the I. ricinus tick models, individual burdens of larvae or nymphs were also added as explanatory variables. Variables were then removed using a backward stepwise selection process to minimise the Akaike information criterion (AIC). A difference in the AIC value of two between models was used as justification to either retain or remove explanatory variables from the models (table of model selection is available in the supplementary materials). Model fit was further examined by performing a likelihood-ratio test between each pair of nested models. Poisson GLMMs were examined for overdispersion and all models were validated by examining diagnostic plots of Pearson and deviance residuals against fitted values and the explanatory variables that were retained in the final model. The models were further simplified by replacing the Tlr4 variable (that included all three genotypes) with Tlr4-1, indicating whether the Tlr4-1 allele was present or absent and repeating the modelling process. In addition, repeat observations were removed and the modelling process was repeated using generalised linear models (GLMs). In all cases, the outcome of the final model was the same for GLMs and GLMMs.
For the models that showed a significant effect of Tlr4 genotype on parasite abundance, individual parasite burden was predicted using the lsmeans function (i.e. least-squares means for specified factors or factor combinations in a linear model) from the lsmeans package v 2.12 (Lenth 2014). Predictions were performed for adult males and females and juvenile males and females in the month of May. All other experimental variables were kept constant at an average value.
Results
A total of 280 individual water voles were genotyped at the Tlr4 locus. The population sizes reported here represent 93 % of the total May 2004 population and 89 % of the total September 2004 population. The discrepancies between the May and September 2004 population numbers reported here and in Oliver et al. 2009a, b are a result of exhausted or degraded DNA. Two functional alleles (Tlr4-1 and Tlr4-2; EMBL accession numbers KJ881164 and KJ881165 respectively) and all three possible genotypes were identified in the population. The two Tlr4 alleles differ by four variable nucleotide positions, of which two of the positions were non-synonymous, and all within the extracellular domain (Fornusková et al. 2013). The overall nucleotide diversity (π) calculated using a Jukes-Cantor correction was 0.005 and the average number of nucleotide differences was four with a stochastic variance under no recombination (Vst) of 2.667. The rate of non-synonymous substitution per non-synonymous site did not significantly exceed the rate of synonymous substitutions per synonymous site (dN = 0.003, dS = 0.012; Z-test = −0.982, p = 0.328). This is inconsistent with the expected effects of balancing selection where dN is expected to be significantly greater than dS.
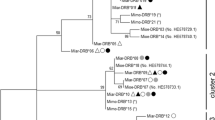
The phylogenetic relationships between the two Tlr4 allele sequences resolved from A. amphibius in this study together with allelic homologues from field vole (Microtus agrestis), bank vole (Myodes glareolus), chestnut white-bellied rat (Niviventer fulvescens), Indochinese forest rat (Rattus andamanensis), red spiny rat (Maxomys surifer), long-tailed giant rat (Leopoldamys neilli), fawn coloured mouse (Mus cervicolor), ryukyo mouse (Mus caroli), house mouse (Mus musculus) and spotted lemur (Lepilemur edwardsi) are shown in Fig. 1. Each species formed a reciprocally monophyletic clade with ≥99 % bootstrap support.

Maximum likelihood phylogeny for the two A. amphibius Tlr4 alleles, together with 30 Tlr4 homologues from ten other species. The underlying model of evolution was identified using Modeltest (Posada and Crandall 1998) as the Tamura 3-parameter model (Tamura 1992) incorporating a discrete Gamma distribution (+G, parameter = 0.3642). Bootstrap support indices are given at nodes (10,000 iterations)
A total of 118 individuals were genotyped from September 2004, which was before the bottleneck. The numbers of water voles was reduced to six individuals (one juvenile, two pregnant females and three males) in 2005 (Oliver and Piertney 2012), then recovered to approximately 70 individuals in 2006 (Fig. 2a). Before the bottleneck the population displayed an excess of heterozygote genotypes relative to Hardy–Weinberg expectations and significant departures from neutrality based on the Ewens–Watterson test (p > 0.05) (Table 1). During and after the bottleneck the population genotype frequencies conformed to Hardy–Weinberg and neutral expectations (Table 1).

Changes in the numbers of voles genotyped (a), Tlr4 functional allele frequencies (b) and genotype frequencies (c) between May 2004 and October 2006
The results from both the pre-bottleneck population simulation and the individual-based simulation indicated that the probability of observing an allele frequency difference of ≤0.04 was very low (0.035 and 0.032 respectively). In May 2004 the population deviated from Hardy–Weinberg expectations due to an excess of heterozygotes (Table 1; Fig. 2c). However, the results from the individual-based simulation indicate that this is unlikely to occur by chance as there was a low probability of the final generation deviating from Hardy–Weinberg expectations (0.038).
In October 2006, when the population had recovered back to 70 individuals, the observed allele frequencies had a difference of 0.32 (0.34 and 0.66 for Tlr4-1 and Tlr4-2 respectively; Fig. 2b). Both the population and the individual-based bottleneck simulations indicated that there was a high probability of observing a final allele frequency difference of ≤0.32 by chance (0.611 and 0.630 respectively). In addition, in October 2006 the population was within Hardy–Weinberg equilibrium which corresponds to the results from the individual-based simulation where the probability of the final generation conforming to Hardy–Weinberg expectations was very high (0.925).
No significant difference in individual burdens of C. nobilis fleas, I. ricinus ticks nymphs or Bartonella was observed between Tlr4 genotypes. The final models implemented for each of these parasite types were as follows: (1) C. nobilis fleas ~ month + reproductive status + year (observations = 194, groups = 173; Poisson error structure), (2) I. ricinus nymphs ~ month + year + I. ricinus larvae burden (observations = 277, groups = 250; Poisson error structure), (3) Bartonella ~ reproductive state + month (observations = 159, groups = 154; Binomial error structure).
However, there was a significant effect of Tlr4 genotype on the burden of M. walkeri fleas, I. ricinus tick larvae and gamasidae mites (model selection results detailed in supplementary material). Individual burdens of M. walkeri varied between 0 and 15 but the majority of voles (94 %) had less than five. The final model was: M. walkeri ~ Tlr4 genotype + reproductive status + sex + month + year (observations = 192, groups = 171; Poisson error structure). The coefficients for this model, including both the Tlr4 terms, are summarised in Table 2. An effect of Tlr4 was observed with adult individuals that had the Tlr4-1 allele in the heterozygote state having significantly fewer M. walkeri than Tlr4-2 homozygote individuals (Table 2; Fig. 3). Male voles had higher M. walkeri burdens than female voles and reproductively inactive voles had higher burdens than individuals that were reproductively active. A seasonal effect was also detected where M. walkeri burdens were higher in May than in October. The individual burdens of M. walkeri fleas predicted from the final model are shown in Fig. 3.
Individual burdens of I. ricinus larvae ranged between 0 and 19 but most voles (95 %) had less than five. The final model was: larvae ~ Tlr4 genotype + reproductive status + month + year + number of I. ricinus nymphs (observations = 277, groups = 250). Individuals with the Tlr4-1 allele in the homozygote form had significantly fewer I. ricinus larvae than heterozygote individuals and individuals that did not have the Tlr4-1 allele present (Table 2; Fig. 3). Reproductively inactive voles had higher burdens than those that were reproductively active and there was a positive relationship found between the numbers of I. ricinus larvae and nymphs. The seasonal effect was also significant with fewer larvae present in September compared to May (Table 2).

Predicted individual burdens for three types of parasite: the flea M. walkeri for a adult males and females and d juvenile males and females, the larvae of the tick I. ricinus for b adult males and females and e juvenile males and females and gamasid mites (Gamasidae) for c adult males and females and f juvenile males and females, according to the genotype at the Tlr4 loci (where Tlr4 genotype 1 = Tlr4-1 homozygote, 2 = heterozygote and 3 = Tlr4-2 homozygote). Predictions were modelled for males and females and adults and juveniles in the month of May. All other significant covariates were held constant at an average value. Bars represent ± epre SE
Individual mite burdens varied between 0 and 208. The final model was: mites ~ Tlr4 genotype + reproductive status + sex + month + year (observations = 142, groups = 121; Poisson error structure). Individuals with the Tlr4-2 allele in the homozygote form had significantly fewer mites than individuals that had the Tlr4-1 allele present (Table 2; Fig. 3). Male voles had a higher burden of mites than females, and reproductively inactive voles had a higher burden than reproductively active voles. Again, a seasonal effect was detected with more mites recorded in May than September (Table 2).
Discussion
Previous studies have demonstrated the role of balancing selection in maintaining TLR diversity (Ferrer-Admetlla et al. 2008; Fisher et al. 2011). The results observed in the present study provide mixed evidence that this is the case for the water vole population on Coiresa. Balancing selection is expected to produce an excess of non-synonymous mutations relative to synonymous mutations across the Tlr4 alleles, and predicted to render para- or polyphyletic patterns of alleles among different species in a phylogeny. Neither of these were apparent in the Coiresa water vole population. The rate of non-synonymous to synonymous mutations across the Tlr4 alleles was not significantly different, and the water vole Tlr4 sequences formed a monophyletic clade with Tlr4 homologues from other species.
Conversely, however, before the bottleneckthe frequencies of the two alleles were closer to parity than would be expected under neutral theory for an island population maintained over long periods at pre-bottleneck densities. Simulation studies highlighted that allele frequencies should show more difference because of the effects of random genetic drift than were actually observed. This suggests that balancing selection is operating to maintain both alleles within the population. The parameters utilised in the simulations favoured the effects of drift in that the duration of the model was 100 generations and population size was kept constant at around the carrying capacity of the island. In reality, the population will have been isolated for much longer than this, and would have been subjected to demographic perturbation resulting in occasional reductions in population size. As such the argument that balancing selection is operating in the Coiresa population is actually more compelling than the test statistics would suggest. Moreover, the population displayed a significant excess of heterozygote genotypes relative to Hardy–Weinberg expectations and allele frequencies departed from neutral expectations. Both of these patterns would be expected if balancing selection was operating.
Interestingly, however, balancing selection was not invoked in the dynamics of the two alleles through the population bottleneck. Ewens–Watterson tests indicated that the changes in allele frequency through the bottleneck were consistent with neutral prediction, with the simulation studies highlighting that the observed disparity in allele frequency between the Tlr4-1 and Tlr4-2 alleles in October 2006 was no more or less than would be expected by chance.
Despite the dramatic reduction in population size during the bottleneck (118 in 2004 to 6 in 2005) the relative brevity of the bottleneck meant that genetic drift did not cause extinction of one of the alleles. A longer duration covering more generations may well have meant that one of the alleles would have been lost with an overall reduction in allelic diversity. The maintenance of both alleles through the bottleneck was also favourably affected by the near parity of allele frequencies at the onset of the population crash. As such balancing selection provided a buffering capacity against drift through the bottleneck. If the starting allele frequencies were more skewed then our simulation studies highlighted a greater propensity for extinction. For example, if we change our simulation parameters from the observed starting allele frequencies to a scenario where one is 0.9 and the other is 0.1 then extinction of the minor occurred in more than 50 % of the replicates.
It has been demonstrated in loci such as the MHC that balancing selection can be mediated through host-parasite interaction, with differences between genotypes and parasite load (Altizer et al. 2003; Prugnolle et al. 2005; Zueva et al. 2014). Indeed, within this population of water voles, MHC variation explained significant variance in gamasid mites (Gamasidae), M. walkeri fleas, I. ricinus tick nymphs and overall co-infection (Oliver et al. 2009a, b). Here, we undertook an equivalent examination where individual Tlr4 genotype was shown to have a significant effect on burdens of M. walker fleas, I.ricinus tick larvae and mites (Gamasidae). Individuals that carried two copies of the Tlr4-2 allele had significantly lower mite burdens than those that did not. Conversely, individuals with two copies of the Tlr4-1 allele had significantly lower burdens of I. ricinus larvae than the heterozygote or Tlr4-2 homozygote equivalents. Lastly, adult individuals with the Tlr4-1 allele in the heterozygote form had fewer M. walkeri fleas than the Tlr4-2 homozygotes.
The results of the mite and I. ricinus larvae analysis show the effects of a specific allele in a homozygote form resulting in a significantly lower burden of parasites, whereas the M. walkeri analysis demonstrated that only one copy of the Tlr4-1 allele was necessary to result in a lower flea burden. Although separately these models suggest evidence of different modes of selection, the overall fitness of an individual will be dictated by its ability to defend against a range of immunological assaults and, when this analysis is considered as a whole, it appears there is an overall Tlr4 heterozygote advantage. This would explain the excess of heterozygote individuals observed prior to the population bottleneck.
For M. walkeri fleas and gamisid mites males had a higher parasite load than females. Male-biased parasitism rates have previously been reported in a number of rodent species. However, the underlying mechanisms through which this is mediated remain unclear (Krasnov et al. 2005; Ferrari et al. 2007; Matthee et al. 2010). The potential effects of testosterone causing immunocompromise are difficult to reconcile with our observation, reproductively inactive individuals had a higher parasite burden than reproductively active individuals. The latter could be the result of parasites favouring the weaker and less resistant juvenile voles as they are easier to infect and exploit (Christe et al. 1998; Hawlena et al. 2005).
The relationship between Tlr4 and various ectoparasite species has previously been demonstrated across a range of different host species (Maizels 2009; Zhang et al. 2011; Brake and Pèrez de León 2012; Wells et al. 2012). The identification of specific signalling pathways during the host’s response to ectoparasites has highlighted the role Tlr4 plays in the innate immune response. Most ectoparasite species, including fleas (Robinson et al. 2008; Matsuura 2013), ticks (Stojek and Dutkiewicz 2004; Zhao et al. 2013; Brannan et al. 2014) and mites (Kim et al. 2014) release infectious agents and potent immunogens in their saliva to downregulate the host’s immune defences. This stimulates the host’s immunoregulatory and effector responses, thus initiating the immune response (Wikel 1999). Tlr4 is therefore one of the first lines of defence against infection and an important link between pathogen recognition and the activation of the immune response (Wikel 1999). Tlr4 plays an important role in activating the immune response by initially detecting the lipopolysaccharide cell wall component of the gram-negative bacterium released by the parasite and initiating the innate immune response, resulting in an inflammatory response and the activation of naïve T-cells by antigen presenting cells (Mogensen 2009). There are many different strains of gram negative bacteria that may be transmitted by parasite species, common strains that may be carried by ticks, fleas and/or mites include bacteria from the genus Borrelia (Rijpkema et al. 2009) and Bartonella (Billiter et al. 2008), although there are many other potential gram negative bacterium’s. Variation within the pathogen-recognition extracellular domain of the Tlr4 gene, most likely maintained by variations in TLR ligands among pathogens, allows for a range of different gram negative bacterium’s to de detected (Maizels 2009). If an individual’s ability to detect and defend against invading infectious agents is compromised or limited, the activation of the acquired immune system in hindered and the individuals parasite burden is likely to increase.
Given that Tlr4 had a significant effect on M. walkeri fleas and I. ricinus tick larvae burdens it is unclear why we don’t see an equivalent pattern in C. nobilis fleas or I. ricinus nymphs. This may reflect statistical power, but given the same discrepancy between the flea species was also apparent with the MHC it may also reflect differences in their individual pathologies. The absence of linkage equilibrium between the MHC and Tlr4 loci indicates that these patterns are independent and not caused by epistatic interaction (Lewontin 1963). In addition, it is surprising that Tlr4 (in either the genotype or allele specific form) was not significant in the Bartonella model given that Bartonella is a gram-negative bacterium. However, unlike the ectoparasite species, blood samples were only collected in the May 2004 and September 2004 sampling sessions. Therefore, the overall sample size for Bartonella is comparatively smaller, reducing the statistical power of any test. Furthermore, as Bartonella was recorded in the blood samples as either present of absent we were unable to account for any variation in the severity of infection between individuals.
What is apparent from this study is that Tlr4 is an important component of the immunogenomic response to pathogens that explains a significant proportion of the variation in individual parasite load. This may have potential consequences for survival, fitness, fecundity and reproductive success of water voles and consequently population viability and persistence. As such, it is an ecologically meaningful locus and understanding its dynamics in populations is useful in a conservation context (Hedrick 2001; Seddon 2010; Thomas 2011; Oliver and Piertney 2012; Grueber et al. 2013). This reinforces the point made by Acevedo-Whitehouse and Cunningham (2006) that the MHC is not the only region of the immunogenome that is important and should be characterised in an ecological context.
The advent of approaches for assaying diversity across the genome in non-model species provides opportunities for identifying other loci that display disproportionately high allelic variation or heterozygosity despite the effects of stochastic microevolutionary processes operating to drive down diversity at the remainder of the genome, or that display high rates of non-synonymous mutations per non-synonymous site. This will circumvent the need for “bottom-up” candidate gene approaches (Piertney and Webster 2008), and no doubt identify other genes that will add to the portfolio of ecologically meaningful adaptive markers such as Tlr4 and MHC.
References
Acevedo-Whitehouse K, Cunningham AA (2006) Is MHC enough for understanding wildlife immunogenetics? Trends Ecol Evol 21:433–438
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2:675–680
Alcaide M, Edwards SV (2011) Molecular evolution of the toll-like receptor multigene family in birds. Mol Biol Evol 28:1703–1715
Allendorf FW (1986) Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol 5:181–190
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709
Altizer S, Harvell D, Friedle E (2003) Rapid evolutionary dynamics and disease threats to biodiversity. Trends Ecol Evol 18:589–596
Bassam BJ, Gresshoff PM (2007) Silver staining DNA in polyacrylamide gels. Nat Protoc 2:2649–2654
Bates D, Maechler M, Bolker B, Walker S (2014) lme4: linear mixed-effects models using Eigen and S4. R package version 1.1-5. http://CRAN.R-project.org/package=lme4
Bergman IM, Edman K, Ekdahl KN, Rosengren KJ, Edfors I (2012) Extensive polymorphism in the porcine Toll-like receptor 10 gene. Int J Immunogenet 39:68–76
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Billiter S, Levy M, Chomel B, Breitschwerdt E (2008) Vector transmission of Bartonella species with emphasis on the potential for tick transmission. Med Vet Entomol 22(1):1–15
Blackwell JM (2001) Genetics and genomics in infectious disease susceptibility. Trends Mol Med 7:521–526
Bollmer JL, Ruder EA, Johnson JA, Eimes JA, Dunn PO (2011) Drift and selection influence geographic variation at immune loci of prairie-chickens. Mol Ecol 20:4695–4706
Brake DK, Pèrez de León AA (2012) Immunoregulation of bovine macrophages by factors in the salivary glands of Rhipicephalus microplus. Parasites Vectors 5(1):38
Brannan JL, Riggs PK, Olafson PU, Ivanov I, Holman PJ (2014) Expression of bovine genes associated with local and systemic immune response to infestation with the lone star tick, Amblyomma americanum. Ticks Tick-Borne Dis 5(6):676–688
Brownlie R, Allan B (2011) Avian toll-like receptors. Cell Tissue Res 343:121–130
Charlesworth D, Charlesworth B (1987) Inbreeding depression and its evolutionary consequences. Annu Rev Ecol Syst 18:237–268
Christe P, Møller AP, de Lope F (1998) Immunocompetence and nestling survival in the house martin: the tasty chick hypothesis. Oikos 83:175–179
Consuegra S, Garcia de Leaniz C (2008) MHC-mediated mate choice increases parasite resistance in salmon. Proc R Soc B 275:1397–1403
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Dunn PO, Bollmer JL, Freeman-Gallant CR, Whittingham LA (2013) MHC variation is related to a sexually selected ornament, survival, and parasite resistance in common yellowthroats. Evolution 67:679–687
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Ferrari N, Rosá R, Pugliese A, Hudson P (2007) The role of sex in parasite dynamics: model simulations of transmission of Heligmosomoides polygyrus in populations of yellow-necked mice, Apodemus flavicollis. Int J Parasitol 37(3–4):341–349
Ferrer-Admetlla A, Bosch E, Sikora M et al (2008) Balancing selection is the main force shaping the evolution of innate immunity genes. J Immunol 181:1315–1322
Fisher CA, Bhattarai EK, Osterstock JB et al (2011) Evolution of the Bovine TLR gene family and member associations with Mycobacterium avium subspecies paratuberculosis infection. PLoS One 6(11):e27744
Fornůsková A, Vinkler M, Pagès M, Galan M, Jousselin E, Cerqueira F, Morand S, Charbonnel Bryja J, Cosson JF (2013) Contrasted evolutionary histories of two toll-like receptors (Tlr4 and Tlr7) in wild rodents (MURINAE). BMC Evol Biol 13:1471–2148
Garantziotis S, Hollingsworth JW, Zaas AK, Schwartz DA (2008) The effect of toll-like receptors and toll-like receptor genetics in human disease. Annu Rev Med 59:343–359
Gay NJ, Gangloff M (2007) Structure and function of toll receptors and their ligands. Annu Rev Biochem 76:141–165
Goudet J (2005) FSTAT: a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). http://www2.unil.ch/popgen/softwares/fstat.htm
Grueber CE, Wallis GP, King TM, Jamieson IG (2012) Variation at innate immunity toll-like receptor genes in a bottlenecked population of a New Zealand robin. PLoS One 7:e45011
Grueber CE, Wallis GP, Jamieson IG (2013) Genetic drift outweighs natural selection at toll-like receptor (TLR) immunity loci in a re-introduced population of a threatened species. Mol Ecol 22:4470–4482
Hawlena H, Abramsky Z, Krasnov BR (2005) Age-based parasitism and density-dependant distribution of fleas (Siphonaptera) on a desert rodent. Popul Ecol 146:200–208
Hedrick PW (2001) Conservation genetics: where are we now? Trends Ecol Evol 16:629–636
Horner DS, Pavesi G, Casrignano T, De Meo PD, Liuni S, Sammeth M, Picardi E, Pesole G (2010) Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief Bioinform 11:181–197
Hughes AL, Friedman R (2008) Codon-based tests of positive selection, branch lengths, and the evolution of mammalian immune system genes. Immunogenetics 60:495–506
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Kawai T, Akira S (2005) Pathogen recognition with Toll-like receptors. Curr Opin Immunol 17:338–344
Keller LF, Jeffery KJM, Arcese P, Beaumont MA, Hochachka WM, Smith JNM, Bruford MW (2001) Immigration and the ephemerality of a natural population bottleneck: evidence form molecular markers. Proc R Soc B 268:1387–1394
Kim EH, Lee JS, Lee NR, Baek SY, Kim EJ, Lee SJ, Kim IS (2014) Regulation of constitutive neutrophil apoptosis due to house dust mite allergen in normal and allergic rhinitis subjects. PLoS One 9(9), article number: e105814
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, New York
Krasnov B, Morand S, Hawlena H, Khokhlova I, Shenbrot G (2005) Sex-biased parasitism, seasonality and sexual size dimorphism in desert rodents. Oecologia 146(2):209–217
Lenth RV (2014). lsmeans: least-squares means. R package version 2.12. http://CRAN.R-project.org/package=lsmeans
Lewontin RC (1963) The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49:49–67
Librado P, Rozas J (2009) DNASP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Maizels RM (2009) Parasite immunomodulation and polymorphisms of the immune system. J Biol 8:61–64
Matsuura M (2013) Structural modifications of bacterial lipopolysaccharide that facilitate gram-negative bacteria evasion of host innate immunity. Front Immunol. doi:10.3389/fimmu.2013.00109
Matthee S, McGeoch M, Krasnov B (2010) Parasite-specific variation and the extent of male-biased parasitism; an example with a South African rodent and ectoparasitic arthropods. Parasitology 137(4):651–660
Merila J, Crnokrak P (2001) Comparison of genetic differentiation at marker loci and quantitative traits. J Evol Biol 14(6):892–903
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22(2):240–273
Mullenbach R, Lagoda PJ, Welter C (1989) An efficient salt-chloroform extraction of DNA from blood and tissues. Trends Genet 5:391
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Oliver MK, Piertney SB (2012) Selection maintains MHC diversity through a natural population bottleneck. Mol Biol Evol 29:1713–1720
Oliver MK, Lambin X, Cornulier T, Piertney SB (2009a) Spatio-temporal variation in the strength and mode of selection acting on major histocompatibility complex diversity in water vole (Arvicola amphibius) metapopulations. Mol Ecol 18:80–92
Oliver MK, Telfer S, Piertney SB (2009b) Major histocompatibility complex (MHC) heterozygote superiority to natural multi-parasite infections in the water vole (Arvicola amphibius). Proc R Soc B 276:1119–1128
Ouborg NJ, Pertoldi C, Loeschcke V, Bijlsma RK, Hedrick PW (2010) Conservation genetics in transition to conservation genomics. Trends Genet 26:177–187
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21
Piertney SB, Webster LMI (2008) Characterising functionally important and ecologically meaningful genetic diversity using a candidate gene approach. Genetica 138(4):419–432
Posada D, Crandall KA (1998) MODEL TEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Prugnolle F, Manica A, Charpentier M, Guégan JF, Guernier V, Balloux F (2005) Pathogen-driven selection and worldwide HLA Class I diversity. Curr Biol 15:1022–1027
Reed DH, Frankham R (2001) How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution 55:1095–1103
Ridley M (2003) Evolution, 3rd edn. Blackwell, Oxford
Rijpkema S, Herbes R, Verbeek-De Kruif N, Schellekens J (2009) Detection of four species of Borrelia burgdorferi sensu lato in Ixodes ricinus ticks collected from roe deer (Capreolus capreolus) in the Netherlands. Epidemiol Infect 117(3):563
Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A (2005) The evolution of vertebrate toll-like receptors. Proc Natl Acad Sci USA 102:9577–9582
Robinson RT, Khader SA, Locksley RM, Lien E, Smiley ST, Cooper AM (2008) Yersinia pestis evades TLR4-dependent induction of IL-12(p40)2 by dendritic cells and subsequent cell migration. J Immunol 181(8):5560–5567
Rousset F (2008) Genepop’007: a complete reimplementation of the Genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
R Core Team (2013). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Seddon PJ (2010) From reintroduaction to assisted colonisation: moving along the conservation translocation spectrum. Restor Ecol 18:796–802
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. Natl Cent Biotechnol Inf 116:3015–3025
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16. doi:10.1186/1742-9994-2-16
Stewart WA, Piertney SB, Dallas JF (1998) Isolation and characterisation of highly polymorphic microsatellites in the water vole, Arvicola terrestris. Mol Ecol 7:1258–1259
Stojek NM, Dutkiewicz J (2004) Studies on the occurrence of gram-negative bacteria in ticks: ixodes ricinus as a potential vector of Pasteurella. Annu Agric Environ Med 2:319–322
Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S (1999) Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451
Tamura K (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol 9:678–687
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA 6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Taylor SS, Jenkins DA, Arcese P (2012) Loss of MHC and neutral variation in Peary caribou: genetic drift is not mitigated by by balancing selection exacerbated by MHC allele distributions. PLoS One 7:e36748
Telfer S, Dallas JF, Aars J, Piertney SB, Stewart WA, Lambin X (2003) Demographic and genetic structure of fossorial water voles (Arvicola terrestris) on Scottish islands. J Zool 259:23–29
Thomas CD (2011) Translocation of species, climate change, and the end of trying to recreate past ecological communities. Trends Ecol Evol 26:216–221
Tschirren B, Raberg L, Westerdahl H (2011) Signatures of selection acting on the innate immunity gene toll-like receptor 2 (TLR2) during the evolutionary history of rodents. J Evol Biol 24:1232–1240
Tschirren B, Andersson M, Scherman K, Westerdahl H, Mittl PRE, Raberg L (2013) Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proc R Soc B 280:20130364. doi:10.1098/rspb.2013.0364
Turner AK, Begon M, Jackson JA, Paterson S (2012) Evidence for selection at cytokine loci in a natural population of field voles (Microtus agrestis). Mol Ecol 21:1632–1646
Uematsu S, Akira S (2008) Toll-like receptors (TLRs) and their ligands: handbook of experimental pharmacology, vol 183. Springer, Berlin, pp 1–20
Watterson GA (1978) The homozygosity test of neutrality. Genetics 88:405–417
Wells B, Burgess ST, McNeilly TN, Huntley JF, Nisbet AJ (2012) Recent developments in the diagnosis of ectoparasite infections and disease through a better understanding of parasite biology and host responses. Mol Cell Probes 26(1):47–53
Werling D, Jann OC, Offord V, Glass EJ, Coffey TJ (2009) Variation matters: TLR structure and species-specific pathogen recognition. Trends Immunol 30(3):124–130
Wikel SK (1999) Modulation of the host immune system by ectoparasite arthropods. Bioscience 49(4):311
Wisely SM, Buskirk SW, Fleming MA, McDonald DB, Ostrander EA (2002) Genetic diversity and fitness in black-footed ferrets before and during a bottleneck. J Hered 93:231–237
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Zhang M, Gao Y, Du X, Zhang D, Ji M, Wu G (2011) Toll-like receptor (TLR) 2 and TLR4 deficiencies exert differential in vivo effects against Schistosomal japonicum. Parasite Immunol 33(4):199–209
Zhao G, Yu M, Cui QW, Zhou JC, Li HX, Qu KX, Wang GL, Huang BZ (2013) Association of bovine toll-like receptor 4 with tick infestation rates and blood histamine concentration. Genet Mol Res 12(3):2783–2793
Zueva KJ, Lumme J, Veselov AE, Kent MP, Lien S, Primmer CR (2014) Footprints of directional selection in wild Atlantic salmon populations: evidence for parasite-driven evolution? PLoS One. doi:10.1371/journal.pone.0091672
Acknowledgments
We would like to thank all colleagues who have contributed to fieldwork and sampling during this study. We would especially like to thank Marius Wenzel and Sandra Telfer for collaboration with different aspects of the study, and Dave Jones and Nat Jones for Bartonella PCR assays. This work was supported by the BBSRC studentship to MKG (BB/J01446X/1) and a NERC studentship to MKO. The research was carried out under project license PPL 40/1813.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gavan, M.K., Oliver, M.K., Douglas, A. et al. Gene dynamics of toll-like receptor 4 through a population bottleneck in an insular population of water voles (Arvicola amphibius). Conserv Genet 16, 1181–1193 (2015). https://doi.org/10.1007/s10592-015-0731-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-015-0731-3