
Abstract
KISS1, a metastasis suppressor gene, has been shown to block metastasis without affecting primary tumor formation. Loss of KISS1 leads to invasion and metastasis in multiple cancers, which is the leading cause of cancer morbidity and mortality. The discovery of KISS1 has provided a ray of hope for early clinical diagnosis and for designing effective treatments targeting metastatic cancer. However, this goal requires greater holistic understanding of its mechanism of action. In this review, we go back into history and highlight some key developments, from the discovery of KISS1 to its role in regulating multiple physiological processes including cancer. We discuss key emerging roles for KISS1, specifically interactions with tissue microenvironment to promote dormancy and regulation of tumor cell metabolism, acknowledged as some of the key players in tumor progression and metastasis. We finally discuss strategies whereby KISS1 might be exploited clinically to treat metastasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
KISS1, originally discovered as a metastasis suppressor, has now burgeoned to having roles as diverse as regulating fertility, reproduction, metabolism and social behaviour [1,2,3,4,5,6,7]. It all started as an exploratory study in 1996, when our laboratory, then based at the Pennsylvania State University College of Medicine in Hershey, set about identifying gene(s) that block metastasis in melanoma cells. Deletions and rearrangements on chromosome 6 associated with melanoma progression and metastasis, compelled introduction of full-length chromosome 6 in metastatic melanoma cell lines using microcell-mediated chromosome transfer [8]. The resulting hybrids were suppressed for metastasis without blocking primary tumor growth. These observations and subsequent studies—which are discussed in subsequent sections—laid the foundation to the discovery of KISS1 [9, 10]. The name KISS1 was a tribute to the place it was discovered—the home of Hershey’s chocolate “Kisses”. Studies have shown loss of KISS1 to be associated with tumor progression and metastasis in multiple but not all cancers [11,12,13].
Shortly thereafter, an orphan G-protein coupled receptor (GPCR) sharing significant sequence homology with galanin receptors was identified in rats and was named GPR54 [14]. In 2001, a significant breakthrough changed the face of KISS1 biology, three labs independently showed that KISS1 is cleaved to smaller peptides of different molecular weights and GPR54 was the natural receptor for these peptides [15,16,17]. The KISS1-derived peptides were named kisspeptins (KP)—KP54, 14, 13 and 10 depending on the number of amino acids in the peptide chain. Among these, KP54 was originally named metastin, a reflection on its anti-metastatic function [18]. With subsequent discoveries attributing a myriad of other functions to metastin, the name KP54 has now become the generally accepted standard nomenclature.
Interestingly, KP are not the only peptide products derived from KISS1. KP10 is further cleaved by matrix metalloproteinases (MMP) resulting in removal of the C-terminal amidated end containing the tripeptide Leu-Arg-Phe-NH2 [19]. This new cleavage product is termed as kissorphin (KSO). KSO can be further modified by amidation at the C-terminus, a characteristic of many bioactive neuropeptides [20]. KSO shares sequence similarities with neuropeptide FF (NPFF) and activates NPFF receptors. KSO inhibits forskolin-mediated release of cyclic adenosine monophosphate (cAMP) and possibly have neuroprotective roles [21, 22]. KSO does not interact with nor activate KISS1R signaling pathways involved in release of gonadotrophins. The identification of KSO suggest the probability of the presence of additional KISS1-derived peptides, some of which will be of equal size but derived from different regions of KISS1 protein (Table 1 lists the nomenclature of known KISS1 derived peptides). We therefore recommend a naming convention, with all KISS1-derived peptides termed as KP irrespective of their size or the method of generation. We also recommend naming KP by position, not size, to reduce confusion in the future. Nonetheless, in this review, we will utilize current nomenclature to minimize confusion.
Establishment of a consensus naming convention has already been adopted for the so-called KISS1 receptor. Originally the orphan receptor (GPR54), it is now known as KISS1R to reflect its now-defined function [18]. The KISS1R pathway has been implicated to be one of the routes for mediating the anti-metastatic effect of KISS1 metastasis suppression [11, 12, 23]. Interestingly, these studies concomitantly reported high levels of KISS1 in specific brain tissues and in select other peripheral tissues and organs, e.g., the pancreas, placenta, liver, kidney and the gonads [16, 17, 24, 25]. These unexpected observations laid the foundation for later discoveries on varied physiological roles for KISS1. In this review, we focus on the role of microenvironment in the KISS1/KISS1R system with specific emphasis on its anti-metastatic role.
KISS1 regulation of physiological processes
In 2003 interest in KISS1 peaked when two laboratories independently showed that patients with isolated hypogonadotropic hypogonadism—a condition characterized by reduced levels of pituitary gonadotrophins and steroid hormones leading to failure of puberty and reproductive defects—presented inactivating mutations and deletions of the KISS1R gene [26, 27]. KISS1R knockout mice recapitulated the reproductive defects observed in humans and activating mutations in KISS1R resulted in precocious puberty in both rats and humans [28,29,30]. Furthermore, mice lacking KISS1 displayed a similar phenotype as KISS1R knockout mice with failure or delayed pubertal development and concomitant reproductive defects [28]. The first reported mutations in the human KISS1 gene showed clinical manifestations which broadly mimicked those observed with KISS1R gene deletions/mutations [31, 32]. In summary, KISS1–KISS1R axis is a gate keeper or a permissive signal for puberty and sexual maturation and its disruption leads to sterility and reproductive defects.
KP are now considered critical players in the hypothalamic–pituitary–gonadal (HPG) signaling axis. KP produced by so-called KISS1 neurons in the hypothalamus stimulate gonadotropin-releasing hormone (GnRH) neurons in the hypothalamus by binding to neurons expressing KISS1R. GnRH neurons release GnRH, which stimulates the release of gonadotrophins—follicle stimulating hormone (FSH) and luteinizing hormone (LH)—from the pituitary gonadotropic cells [5, 30]. FSH and LH then regulate the secretion of sex hormones from the ovaries and testes and production of eggs and sperm contributing to onset of puberty and sexual maturation. This has been supported by several studies where exogenous treatment with KP stimulated release of GnRH and treatment with KISS1R antagonist peptide-234 blocked the surge of gonadotrophins [12, 33,34,35,36,37,38].
The KISS1–KISS1R system is not limited to mammals and is present across non-mammalian vertebrates stretching from fishes to reptiles pointing to conserved roles for the KISS signaling [38]. In mammals, only one gene for KISS1 and one for KISS1R have been reported; however, non-mammalian vertebrates have two KISS (KISS1 and KISS2) and four KISS1R coding genes, with some amphibians expressing three forms of the KISS gene [38]. KISS1 and its paralog KISS2 are generally well conserved across species; however, they share minimal amino acid sequence similarity. Different forms of KISS1R, however, share significant sequence homology across species. Synteny analysis shows KISS was duplicated before divergence of sarcopterygians and actinopterygians. The KISS2 paralog was however lost in placental mammals [38,39,40]. Neuronal in situ hybridization in fishes showed high KISS1 mRNA expression in habenula and hypothalamic regions and KISS2 mRNA expression in the posterior tuberal nucleus and hypothalamic regions of the brain [40, 41]. KP derived from KISS1 regulate GnRH levels, albeit with different potencies. KP also control pubertal onset and serve as a link on environmental control of reproductive behavior in fishes. These observations strongly imply a role for KP signaling in regulating reproductive behaviour in non-mammalian vertebrates.
KP also profoundly impact puberty and reproductive behaviour in mammals apart from humans, including rodents, primates and ruminants [42,43,44]. The mechanism of KP secretion and its regulation of puberty varies from species to species. The entire HPG axis is regulated via positive and negative feedback loops influenced by the sex steroids and controlled by multiple environmental and metabolic cues [2, 3, 45]. Interestingly, sex differences influence the distribution and number of KISS1 neurons, with females having many more KISS1 expressing cells [43]. Exposure to different sex hormones during development and sexual maturation possibly contributes to these variations. In female rats and sheep, estrogen signaling plays a key role in release of gonadotrophins by regulating KISS1 expression on arcuate hypothalamus (KISS1ARH). Estrogen strongly suppresses KISS1 release from KISS1ARH neurons during the pre-pubertal period, an increase in KISS1 levels at puberty coincides with loss of inhibitory effect of estrogen on these neurons [44]. However, in primates, estrogen-dependent suppression of gonadotropin secretion occurs just before puberty onset in monkeys. Thus, the central mechanism inhibiting GnRH/gonadotropin secretion in primates appears to differ from that in rodents and sheep [44]. Plasma KP have been observed at very high concentrations in pregnant women reaching 200-fold higher levels in the third trimester [5, 30, 42]. KP have been shown to be key regulators in ovulation, follicular maturation, embryo implantation, inhibiting trophoblast invasion and placental angiogenesis [37, 44, 46, 47].
KISS1 neurons are also critical components of the hypothalamic circadian oscillator network and are central to balancing metabolic requirements and behaviour [1, 45, 48]. KISS1 neurons in the KISS1ARH regulate food intake, metabolic activity and the circadian cycle in mouse models [1]. This observation is critical since reproductive events and pregnancy depend upon substantial energy reserves which require coordinated signaling between neurons controlling food intake and energy levels with neurons regulating reproductive behavior. Loss of KISS1ARH neurons shifts circadian rhythms affecting food consumption, physiological behaviour and pulsatile release of GnRH, contributing to fertility disorders [1]. Results from a recent whole-body knockout of KISS1R in mice resulted in hypogonadism, as expected. However, female, but not male, mice were obese and exhibited adiposity and metabolic shifts [49]. The sex differences highlight how KISS1/KISS1R signaling works in concert with other systems in cells and physiologically to elicit complex traits, some of which will be elaborated below.
KP tend to be higher in type 2 diabetes mellitus (T2DM) patients, where they suppress pancreatic β cell insulin secretion [50]. Interestingly, the antagonistic hormone glucagon inhibits insulin secretion in pancreatic β cells by upregulating hepatic production of KISS1 via the cyclic AMP-Protein Kinase A (cAMP-PKA) pathway, independent of hepatic glucose production and hyperglycaemia [50]. The KP released by hepatocytes directly interacts with KISS1R in the pancreatic islet β-cell, leading to impairs insulin production. Furthermore, circulating KP at nanomolar concentrations inhibits glucose stimulated insulin secretion (GSIS) in β cells. However, KP at higher micromolar concentrations stimulates GSIS, possibly through effects independent of KISS1R indicating a dose-dependent difference in KISS1 regulation of GSIS [50]. KISS1 has also been shown to impact estrogen-dependent skeletal homeostasis and motivational drive for palatable food in females and modulating fear responses in zebrafish suggesting it may be a link between environmental, metabolic events to reproductive behavioural patterns [2, 3, 7, 45, 50]. For a gene family that was first discovered as regulating metastasis, it has transcended well beyond this field having impact on multiple physiological processes that would not have been envisioned at the time of its discovery.
KISS1 as a metastasis suppressor
Metastasis suppressors are a family of genes that inhibit the metastatic spread of cancer without blocking primary tumor formation. Therefore, they are critical regulators of the metastatic process/cascade [51]. Currently, there are over 30 functionally verified metastasis suppressors that block various steps in the metastatic cascade, from tumor cell invasion to colonization, preventing development of metastasis [52, 53]. The mechanisms of action of many of these genes are still largely unknown. If metastasis suppressor functions could be translated into clinical use, they could increase survival and quality of life for many cancer patients [51, 54, 55].
The KISS1 gene was initially thought to reside on chromosome 6 since that was the genetic defect that was repaired by microcell-mediated chromosome transfer. Surprisingly however, KISS1 was mapped on the long arm of chromosome 1 (1q32). Eventually KISS1 was shown to be regulated by protein products of two genes, Thioredoxin Interacting Protein (TXNIP also known as vitamin D Upregulated Protein 1 (VDUP1) or Thioredoxin Binding Protein 2, present on chromosome 1) and Cofactor Required for SP1 activity 3 (CRSP3, also known as vitamin D receptor interacting protein, located on chromosome 6). Indeed, it was CRSP3 that was deleted on chromosome 6 in the first experiments that ultimately led to the discovery of KISS1. Studies have shown CRSP3 indirectly upregulated KISS1 levels by directly modulating TXNIP expression. Loss of CRSP3 was accompanied by reduction in KISS1 expression in melanoma cell lines [9, 24, 56].
Full-length human KISS1 is a 145 amino acid protein with a central hydrophobic core, an N-terminal secretion signal sequence and three pockets of dibasic residues of arginine-lysine (R56-K), arginine-arginine (R66-R) and lysine-arginine (K123-R) interspersed along its length. The calculated size of the protein is around 15.9 kDa. KISS1 was determined as an intrinsically disordered protein lacking secondary/tertiary structure in its entirety, largely extended and without any significant presence of local conformational preferences, making it difficult to predict functional domains within the protein [57,58,59]. Following the discovery of KISS1R and KP, it was acknowledged that KISS1 is cleaved into multiple bioactive peptides and each of these peptides are ligands for KISS1R where they activate or inhibit multiple signaling pathways. Through these discoveries an understanding of the mechanism of action of KISS1 was also inferred. KISS1 is constitutively secreted by cells to the surrounding microenvironment and secretion is essential for its anti-metastatic effect as was demonstrated in studies where tumor cells lacking KISS1 secretion signal sequence were able to successfully metastasize [60]. Interestingly, the processing of KISS1 to KP by the enzyme furin occurs only after secretion, the reasons for which are still not completely clear [61]. KISS1 was also shown to interact extracellularly with proMMP2/9 specifically through cysteine 53 suggesting an involvement of MMPs in KISS1 cleavage [19].
Role and regulation of KISS1 in multiple cancers
KISS1 has been identified as a bona fide metastasis suppressor gene in melanoma, mesothelioma, pancreatic, nasopharyngeal, ovarian, gastric, colorectal, endometrial, bladder, renal, head and neck and esophageal cancers [34, 62,63,64,65,66,67,68,69,70,71,72,73,74,75]. The data on the role of KISS1–KISS1R axis in breast, hepatocellular and thyroid cancer is not conclusive with both pro-and anti-metastatic actions reported [23, 64, 76, 77]; however, there are suggestions that cancer subtype may be a key variable in KISS1 activity. Studies supporting an anti-metastatic role for KISS1 signaling showed KISS1 expression restricted angiogenesis in tumor xenografts of mice with MDA-MB-231 triple negative breast cancer (TNBC) cells, sensitizing them to oncolytic therapy [78]. Furthermore, KISS1 expression reduced brain metastasis in these xenografted mouse models with MDA-MB-231 cells by downregulating pro-angiogenic factors secretion and inhibiting vesicular recycling of epidermal growth factor receptor (EGFR) [79]. KISS1 expression also inhibited melatonin induced invasion in MDA-MB-231 cells [80]. Contrasting these observations, KISS1 and KISS1R expression promoted invadopodia formation in in vitro assays in TNBC MDA-MB-231 and Hs578T cells by inducing EGFR transactivation and increasing ERK1 levels [81,82,83,84]. However, a similar effect of KISS1 was not observed in estrogen and progesterone positive T47D breast carcinoma cells [83]. Similarly, TGFβ, a promoter of metastatic phenotype, strongly induced KISS1 mRNA levels in multiple TNBC cell lines, while having no effect on ER positive cell lines. Furthermore, KISS1 was found to be a direct downstream target of canonical TGFβ/Smad2 pathway and promoted motility, invasion and drug resistance in TNBC cells [85, 86].
Interestingly, estrogen receptor (ER) expression was inversely related to KISS1 levels several breast cancer cell lines, TNBC cells having no ER expression showed moderate to high levels of KISS1 depending on the cell type. MDA-MB-231 cells on introduction of estrogen receptor alpha (ERα) showed a significant reduction in KISS1 expression. Furthermore tamoxifen (TAM) treatment significantly increased KISS1 levels in ERα positive MCF7 and T47D breast cancer cells [83]. However, in breast tumors, both ERα negative and positive, a variable increase in KISS1 expression was observed with tumor grade, complicating the interpretation for a direct link between ER and KISS1 in breast cancer [83, 84]. With a strong ER signaling in breast cancer and in regulation of KISS1 levels, future follow up studies will help draw mechanistic insights on their role in breast cancer metastasis.
Elevated KISS1 levels in hepatocellular carcinoma (HCC) correlated with tumor aggressiveness [33, 87]. However, reports also show KISS1 inhibiting metastasis in HCC by downregulating MMP-9 activity [88]. KISS1 and hypoxia inducible factor-1α (HIF-1α) showed an inverse relationship in HCC, with low KISS1 expression correlating with high HIF-1α levels. Since the role of HIF-1α in promoting tumor angiogenesis and invasion is well known, it is plausible KISS1 may be inhibiting invasion and metastasis by downregulating HIF-1α levels [89]. In thyroid cancers, increased KISS1 expression correlated with tumor invasion to extrathyroidal tissues, however, a decreased expression of KISS1R was tumor progressed was enough to attenuate KISS1 signaling [77, 90]. Future work on the role of the KISS1–KISS1R signaling in these cancers as a whole and an understanding of the molecular mechanisms will possibly help shed light on these contradictory observations.
A pro-metastatic role for KISS1 was observed predominantly in TNBC and HCC. The former is hormone receptor deficient and, perhaps the difference in behavior of TNBC cells may be associated with interactions of KISS1 with hormone-responsive populations. The clinical findings that KISS1 is associated with worse prognosis in some reports for patients with HCC may be associated with KISS1’s associations with metabolism regulation and the liver's central role in body metabolism [45, 48, 50]. Not unexpectedly, KISS1/KISS1R roles in metastasis without blocking primary (orthotopic) tumor growth de facto imply that KISS1/KISS1R are sensors of the environment. It stands to reason, then, that both molecules cooperate with other sensors to alter cellular behavior. Further, it should be anticipated that defects in KISS1–KISS1R axis yield cell- and microenvironment-specific phenotypes. The multifaceted and multi-factorial nature of metastasis regulation must await a more detailed defining of relevant signals.
Multiple microRNA (miR) and long non-coding RNA (lncRNA) regulate KISS1 expression, some acting directly on KISS1 and others indirectly through intermediate players [91]. Induction of miR-345 by CXCL12 produced by astrocytes inhibited KISS1 expression and suppressed metastasis of the brain metastatic breast cancer cell line MDA-MB-231Br [92]. MiR-21 and miR-3648 downregulated transcription factor 21 (TCF21)–KISS1 axis and promoted invasion and metastasis in multiple renal and bladder cancer cell lines [93, 94]. TCF21, a basic helix–loop–helix protein also suppressed invasion and metastasis of human esophageal carcinoma cell line KYSE510 and melanoma cell line C8161 by binding to KISS1 promotor and upregulating its expression [65, 95]. WASF3 a Wiskott-Aldrich cytoskeletal protein was shown to promote invasion and metastasis in MDA-MB-231 breast cancer cells by downregulating KISS1. This released the inhibitory effect of KISS1 on IκBα on NF-κB, which in turn activated pro-invasion genes such as various MMPs and ZEB1 [96, 97]. Activation of ZEB1 led to the downregulation of E-cadherin and upregulation of metastasis promoter miR-200 family of genes [36]. MiR-199b inhibited metastasis of SW620 colorectal carcinoma cell line by directly downregulating the histone deacetylase, sirtuin (SIRT1) leading to the subsequent activation of KISS1 [98]. The long non-coding RNA, LncRNA TP73-AS1 was reported to promote renal carcinoma invasion and metastasis in ccRCC cells by downregulating KISS1 expression [99]. Long noncoding RNA TC0101441 induced epithelial-mesenchymal transition in epithelial ovarian cancer metastasis by downregulating KISS1 levels [100]. Long non-coding RNA MNX1-AS1 promoted osteosarcoma proliferation and invasion via inhibiting KISS1 expression [101]. Similarly, lncRNA LUCAT1 promoted prostate cancer invasion and migration by inhibiting KISS1 expression in PC-3 prostate cancer cells [102].
Mammalian relative of DnaJ long isoform (MRJ(L)) suppressed invasion and motility in MDA-MB-231 and MDA-MB-435 breast cancer cells by upregulating secreted levels of KISS1 [103]. KISS1 was epigenetically silenced in RT4 bladder cancer cell line by Ubiquitin-like with PHD and RING finger domains 1 (UHRF1), resulting in promotion of invasion and metastasis [104]. Interestingly, UHRF1 downregulated peroxisome proliferator-activated receptor gamma (PPARG), a central player in cellular metabolism, connecting KISS1 to cellular metabolism [105].
KISS1 regulation of cellular signaling
Depending upon the cancer type, KISS1 and KP affects signaling through pathways leading to changes in angiogenesis, immune responses, cellular differentiation and proliferation (Table 2 lists the mechanism of KISS1 signaling in multiple cancers). A well-studied signaling pathway is the regulation of intracellular calcium levels and activation of protein kinase C (PKC) [63, 106]. KP binding to KISS1R results in the hydrolysis of phosphatidylinositol (4,5)-bisphosphate (PIP2) by phospholipase C (PLC) leading to the generation of a water-soluble moiety inositol (1,4,5)-trisphosphate (IP3) and the lipid soluble diacylglycerol (DAG). IP3 modulates release of calcium from intracellular stores such as the endoplasmic reticulum regulating the activity of several calcium binding proteins, increasing stress fiber formation, reducing cell motility and chemotaxis and inducing apoptosis [107]. Calcium and DAG also combine to activate PKC, resulting in receptor desensitization, inhibition of epithelial to mesenchymal transition (EMT) by reducing SLUG transcription factor expression, increased E-cadherin expression and activation of protein kinase D1 (PKD1), all associated with the anti-metastatic effect of KISS1 [108, 109].
KISS1 reduced p50/p65 NF-κB interaction with the matrix metalloproteinase 9 (MMP-9) promoter by associating with and preventing degradation of IκB. The dysregulated NF-κB pathway led to decreased MMP-9 levels and reduced migration in MDA-MB 231 and MCF-7 breast cancer cell lines without affecting proliferation in in vitro assays [110]. A similar negative correlation between KISS1 and MMP-9 was also reported in non-small cell lung cancer (NSCLC) patient samples [62, 69]. KP10 treatment suppressed tumor growth in xenografted mice tumor models with MDA-MB-231 breast cancer cells and improved their survival rate [111]. Furthermore, KP10 treatment suppressed intratumoral growth of microvessels suggesting a role for KP10 in inhibiting tumor angiogenesis. This was supported by observations where KP10 administration reduced human umbilical vein endothelial cell (HUVEC) migration, invasion, and tube formation in chick embryo chorioallantoic membrane angiogenesis assay [111]. KP10 administration inhibited VEGF induced phosphorylation of c-Src and focal adhesion kinase (FAK) in human umbilical vein endothelial cell (HUVEC) cells, inactivating Rho GTPases, important mechanisms for promoting angiogenesis, motility and invasion [112, 113]. A similar inhibitory effect of KP10 on tumor angiogenesis was reported in xenografted mice models with PC-3 human prostate cancer cells [114]. KISS1 expression also inhibited invasion in in vitro trans well migration assays in LNCaP and PC-3 prostate cancer cells by reducing the expression levels of Notch 1, an important promoter of angiogenesis [114].
KISS1 activated the mitogen activated protein kinase (MAPK) pathway, increased phosphorylation and activation of p53, triggering of apoptosis in MG-63 osteosarcoma cell line [68, 115]. KP induced the RhoA mediated activation of eukaryotic translation initiation factor 2α kinase 2 (EIF2AK2) which was required for metastasis suppression in human breast SK-BR-3, prostatic PC-3 and colorectal LoVo adenocarcinoma cells [116]. KP10 inhibited chemotaxis in CXCR4 expressing Chinese hamster ovarian (CHO) and HeLa cells in response to ligand CXCL12 leading to reduced invasion through Matrigel [117]. The contradictory actions and the expanding list of signaling events regulated by the KISS1–KISS1R axis makes it sometimes difficult to interpret its mode of action. However, what is apparent is that the KISS1/KISS1R axis regulates diverse signaling cascades which are cell type-specific, possibly influenced by myriad factors such as cellular origin, genetic variations and the characteristics of the tissue microenvironment.
Role of microenvironment in KISS1 induction of dormancy
Tumor cell dormancy, more accurately long remissions followed by recurrence has fascinated clinicians for a long time [118]. With limited knowledge, a spate of lingering questions has emanated such as, how tumor cells become dormant? What factors contribute to dormancy and cell survival? How do the tumor cells awaken from dormancy? And whether keeping cells dormant or therapeutically targeting dormant cells represent the best approach to controlling and treating metastatic disease [119,120,121,122]. Answers to these questions are critical for a thorough understanding of the process and for developing targeted therapeutic approaches targeting these cells.
Cellular dormancy is regulated by a combination of intrinsic and extrinsic factors. Several metastasis suppressors including KISS1 have been shown to induce tumor cell dormancy [53, 123]. Experimental metastasis assays have shown that GFP-labeled KISS1 expressing tumor cells disseminate via the vascular system but do not proliferate at secondary organs. This strongly suggests that KISS1 suppresses metastasis by promoting ectopic dormancy in tumor cells [60, 124]. An intricate balance in ERK/p38 levels plays a critical role in determining the proliferative status of a cell with low ERK/high p38 levels promoting growth arrest and high ERK/low p38 leading to tumor cell proliferation. Several metastasis suppressor genes induce tumor cell dormancy at ectopic sites by increasing p38 activity [119, 125]. Interestingly, the data on KISS1 is not conclusive, overexpression of KISS1 inhibited growth and invasion of MG-63 osteosarcoma cells by decreasing p38 signaling [68]. In stomach cancers, however, KISS1 activates p38 MAPK signaling in NUGC-3 and MKN-28 gastric cancer cells [126]. This indicates variables such as cell type and cellular interaction with the microenvironment or niche are probably key factors influencing the role played by KISS1. Therefore, a holistic analysis is required to understand this critical phenomenon and in vitro assays with artificial environments are insufficient in completely answering these questions.
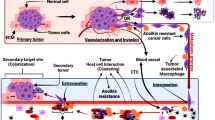
KISS1 also inactivates the phosphoinositide 3-kinase (PI3K)–AKT–mTOR pathway in multiple cancers, possibly contributing to tumor cell quiescence since this axis has been strongly implicated in tumorigenesis and metastasis [127]. ATF6a–Rheb–mTOR signaling is critical to survival of DTCs at ectopic sites, its possible KISS1 not just keeps tumor cells quiescent but also may initiate the process of their destruction. Future studies may provide an answer to this hypothesis. Quiescence is reversible, therefore DTCs may employ various survival means in unfavorable conditions, hold out for microenvironment changes fertile enough to revert back to growth and proliferation [128]. For instance, this may involve activation of autophagic pathways [129]. Autophagy, a cellular process of “self-eating” to tide over unfavorable conditions has been employed by DTCs to survive at ectopic sites [92, 125]. Overexpression of KISS1 in brain metastatic breast cancer cell line MDA-MB-231Br cells reduced ATG5 and ATG7 levels, key inducers of autophagy and inhibited conversion of LC3-I to LC3-II which is critical to autophagosome formation [92]. Therefore, KISS1 overexpression leads to autophagy inhibition in MDA-MB-231Br cells and possibly diminishes their survival chances at ectopic sites. It remains to be seen if KISS1 regulation of autophagy is the means to dormancy and subsequent induction of cell death in DTCs is universal or restricted to certain tumor types. An interesting aspect is that many of the cell lines in which KISS1 suppressed metastasis showed no detectable levels of KISS1R [12, 130]. Furthermore, no anti-metastatic role for KISS1 was observed in C8161.9 melanoma cells that were cultured in artificial environment in a PuMA assay [131]. These observations suggest additional regulatory mechanisms are involved in mediating the anti-metastatic effect of KISS1 possibly involving interaction with the tissue microenvironment or the existence of other receptors to KISS1/KP (Fig. 1).

Possible mechanisms by which KISS1 suppresses metastasis. In in vitro models, KISS1 has been shown to prevent EMT, block angiogenesis by downregulating key angiogenic factors like VEGF and inhibit tumor cell invasion by reducing MMP2 and 9 activity and thus preventing the reorganization of ECM. In in vivo models, KISS1 has been shown to inhibit DTC colonization and keep them in a state of dormancy. The possible mechanism by which KISS1 may induce dormancy may include—autocrine signaling: KISS1/KP could signal through KISS1R or an as-yet unidentified alternative receptor in an autocrine fashion to induce dormancy. Paracrine signaling: KP may activate or induce stromal cells to produce secreted factors that may (in)directly elicit dormancy in DTCs or manipulate the surrounding microenvironment to induce dormancy in DTCs. It is possible both autocrine and non-autocrine mechanisms may exist depending on the tumor cell type and the presence/absence of KISS1R
The presence of KISS1R positive stromal cells and immune cells raises the possibility of some form of communication between tumor cells and surrounding cells of the microenvironment [12]. This may involve possible paracrine signaling between DTCs expressing KISS1 and stromal or immune cells expressing KISS1R. Secreted KISS1/KP may interact with KISS1R on stromal or immune cells of the microenvironment, which then secrete some unknown factor(s) that then interact reciprocally with the tumor cell, keeping it in a state of dormancy. Support for this hypothesis comes from observations in tumor cells expressing truncated version of KISS1, lacking a secretion signal sequence. In these tumor cells, wherein KISS1 protein is expressed but not secreted, the anti-metastatic effect of KISS1 is blocked. Identifying these factor(s) will be crucial in understanding the mechanism of KISS1 mediated quiescence of tumor cells. Interestingly, DTCs share several properties with stem cells, viz. quiescence, activation and proliferation and its plausible they are regulated by similar signals. Multiple factors such as growth-specific arrest protein 6 (GAS6), lysophosphatidic acid (LPA) and transforming growth factor β (TGFβ) family of proteins like bone morphogenetic protein 7 (BMP7), secreted from surrounding osteoblasts and other stromal cells, have been reported to induce dormancy in DTCs [119, 125]. A small population of macrophages has been found to express KISS1R and several studies have implicated tumor associated macrophages and T-lymphocytes in cancer progression, very little is known on their role in promoting DTC dormancy [132, 133]. Whether KISS1 regulates the release of dormancy factors from surrounding cells in the microenvironment or if it has any influence on the immune cells is still not determined.
This reciprocal interaction is also dependent on multiple factors like the organ type where the DTCs reside, type of surrounding cells and their KISS1R expression levels. This is supported by observations wherein, the anti-metastatic effect of KISS1 varies depending on the type of cancer. In some cases, KISS1 also assumes a pro-metastatic role like in hepatocellular carcinoma and breast carcinoma. Taken together, KISS1/KP mainly exerts its anti-metastatic effect by keeping the DTCs in a dormant state. This suggests there is window of opportunity to keep tumor cells in a dormant state using exogenous KISS1/KP therapy and possibly combine this with drug treatments that specifically target dormant cells and eliminate them completely.
KISS1 regulation of the microenvironment through induction of metabolic changes
During the period of dormancy when a tumor cell remains latent, the risk of recurrence depends on changes occurring within both the tumor cell and the surrounding microenvironment. Metabolic changes play a critical role, with quiescent cells in general exhibiting low metabolic rate with more focus on survival. Cancer recurrence is associated with dormant cells becoming active and among many other changes, adapting their metabolism for faster growth and proliferation. These alterations occur in reciprocal cohesion with changes in the microenvironment. They include changes in cellular interactions, pH, recruitment of immune cells and blood vessels, supply of oxygen and changes in cellular biomechanics. Aerobic glycolysis has been shown to be a key feature of tumor metabolism. Tumor cells often exhibit increased uptake glucose and show a preference for glycolysis over oxidative phosphorylation (OXPHOS) for ATP, this has been termed as the “Warburg effect” [134, 135]. The metabolic end products, lactate and pyruvate are secreted and taken up by surrounding tumor cells, a feed forward mechanism for energy production for neighbouring tumor cells to carry out oxidative metabolism. Regaining tumor initiating capabilities is critical for DTCs to successfully establish metastasis at ectopic sites, which they do by adjusting and altering the metabolic environment at these distant sites. Various pathways including glycolysis, OXPHOS, fatty acid and amino acid metabolism have been implicated in cancer metastasis [136].
KISS1 has been shown to play an important role in altering cell metabolism by decreasing aerobic glycolysis and increasing OXPHOS and increasing mitochondrial biogenesis [137]. Interestingly, all these changes were observed with only full-length KISS1 and not with KISS1 that lacks the N-terminal signal peptide, suggesting a possible interplay between the tumor cells and the microenvironment [137]. KISS1 reduces the acidification of extracellular media, reduces glucose uptake and lactate secretion all contributing to reversing the “Warburg effect” in tumor cells. Apart from increasing mitochondrial biogenesis, KISS1 also upregulates several nuclear encoded mitochondrial genes such as the chaperone factors, membrane polarization and small molecule import and export factors. KISS1 also upregulates the expression of two critical transcription factors, mitochondrial nuclear respiratory factors (NRF1) and mitochondrial transcription factor A (Tfam) which are involved in regulating critical mitochondrial genes and in mitochondrial genome replication [137].
KISS1 increased peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) protein levels by protecting it from proteasomal degradation and therefore stabilizing its protein levels [137]. PGC1α, a master regulator of mitochondrial biogenesis controls several metabolic events, fatty acid synthesis and oxidation, progression through citric acid cycle and OXPHOS. In KISS1 expressing tumor cells, PGC1α was found to inhibit fatty acid synthesis and promote beta oxidation through AMP kinase (AMPK) based inhibition of acetyl Co-A carboxylase (ACC), leading to decreased fatty acid synthesis and increased beta oxidation. Furthermore, PGC1α was found to mediate many of KISS1’s metabolic effects and importantly was found to control the anti-metastatic effects of KISS1 since knockdown of PGC1α reversed all the metabolic changes induced by KISS1 and restored a metastatic phenotype. KISS1 also inhibited the mitochondrial hexokinase-II (HKII) leading to increased apoptosis [138]. Taken together, KISS1-expressing cells have increased mitochondrial mass, reduced utilization of glucose and a shift from aerobic glycolysis towards mitochondrial respiration and OXPHOS, increased pH of the extracellular microenvironment possibly due to decreased secretion of lactic acid. Importantly all the changes are reversed when KISS1 secretion to the extracellular environment is blocked. Therefore, a possible manipulation of the microenvironment by KISS1 resulting in altered metabolism of tumor cells at ectopic sites and vice versa represents a potential new connection linking microenvironment, metabolism and establishment of metastasis.
Microenvironment may influence KISS1 anti-metastatic role
It has been assumed that KP interaction with KISS1R was essentially the only mechanism through which KISS1 suppressed metastasis. Initial observations presented an autocrine signaling loop between KISS1/KP ligand and KISS1R receptor. However, of the several cell lines where KISS1 had an anti-metastatic effect, many of them showed no detectable levels of KISS1R expression. This suggests that metastasis suppression by KISS1 does not occur solely via autocrine signaling through KISS1R. It is possible alternate routes exist for KISS1 anti-metastatic function, either through KISS1R expressed in neighbouring stromal cells (paracrine signaling) such as the cancer associated fibroblasts (CAF), immune cells like the macrophages or by KP signaling via a yet unknown receptor. This is further supported by lack of conclusive in vitro assays mimicking in vivo anti-metastatic effect of KISS1 observed in mouse models [131]. A reasonable understanding exists on the KP/KISS1R axis in regulating puberty and reproductive behaviour across species. It is therefore imperative to delineate these “other” pathways to gain a comprehensive understanding of KISS1 function as a metastasis suppressor and in order to develop a broad therapeutic strategy for treatment of metastatic cancers resulting from loss of KISS1 function.
KISS1 a potential therapeutic
The KISS/KP-KISS1R axis has been considered as an attractive therapeutic target, not only in cancers but also for treatment for physiological disturbances due to aberrant KISS1 signaling. This can be attributed to the small size of KP, low immunogenicity and limited side effects. Plasma levels of various KP and KISS1R levels have served as prognostic markers for tumor progression [139]. KISS1 expression increased the sensitivity of head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancers (NSCLC) to cisplatin therapy by mechanistically altering apoptotic and metabolic pathways in response to cisplatin [140]. Both chemically synthesized KP analogs and natural KP have been tested as anti-cancer therapeutics in animal models and in humans for restoring reproductive defects, very much comparable to the use of insulin for diabetics [141]. The results have been promising, KP administration restores near normalcy for reproductive defects in humans and reduces invasion and metastasis in preclinical models of cancers and cell lines [29, 142, 143].
In tumor cells lacking KISS1R but with anti-metastatic effect for KISS1, a role for intact KISS1 cannot be discounted. Viral vehicle-based introduction of metastasis suppressor genes has shown promising results in animal models of cancers, suggesting introduction of full-length KISS1 as an alternative therapeutic strategy. Since expression of KISS1 is regulated by upstream genes TXNIP and CRSP3, modulating their expression may also be a strategy to upregulate KISS1 levels [12]. However, all these options are wrought with their own limitations and a better understanding of the mechanism of action of KISS1 will help design suitable and patient specific therapy. Furthermore, if reciprocal communication between stroma and tumor cells is proven, the stroma will also provide an alternative target for KP based therapy.
Similarly, peripheral delivery of KP stimulates release of gonadotrophins in humans, increasing fertility and pregnancy rates [5, 30]. In pre-pubertal rats circulating levels of gonadotropins increased on central administration of KP [30]. Taking these observations forward, KP54 injection in women showed peak in circulating levels after 40 min of injection [144, 145]. KP10 showed peak detection levels after only 15 min and had a shorter half-life compared to KP54 [146]. The effect of KP on gonadotrophin release was dependent on its type, mode of administration and the stage of ovulation [147]. Furthermore, KP in general stimulated a more pronounced effect on LH release compared to FSH. KP serve as important biomarker for pregnancies, KP levels less than 1630 pmol/L is an indicator for miscarriages [5, 30]. On the other hand, repeated administration of KP resulted in KISS1R desensitization and abrogation of its effects. Variations in potency effect of different KP and differences in their half-life have also been observed [147]. To sum up, KP are promising molecules for therapeutic targeting of fertility disorders and possibly other metabolic and physiological defects, however, significant work remains to be done regarding their route of administration and maintaining their circulating levels and potency for longer periods. Importantly, KISS1/KP administration into people has been successful and safe and has potential as a therapeutic, having already satisfied the first criterion—safety and efficacy for reproductive diseases/syndromes.
Conclusion and future perspective
It has been more than 20 years since the discovery of KISS1, we did not foresee the diverse roles it has been shown to be involved in at the time of its discovery. This is nothing but a victory for explorative science. The progress on the role of KP in reproductive health and fertility has been rapid; however, our knowledge of their contribution to cancer metastasis has comparatively not kept pace. While it’s clear that KISS1 has a largely anti-metastatic role in cancers, a lack of a complete understanding of the signaling mechanisms regulating its anti-metastatic function has hampered the progress in gaining mechanistic insights on KISS1. This is compounded by reports showing several pathways activated by KP-KISS1R axis to have both anti-metastatic and pro-metastatic roles depending on the type of cancer. Several KISS1 expressing cancer cell lines suppressed for metastasis in in vivo experimental assays did not show any detectable KISS1R expression. This raises the possibility of a reciprocal communication between KISS1R expressing stromal cells in the microenvironment and the tumor cells or the possibility of existence of other receptor(s) for KISS1, the answers to both the points is not clear. This is due to research largely being centered on KISS1 the ligand, knowledge on the receptor-KISS1R, its localization and regulation has taken a back seat, mainly due to the lack of good antibodies directed against it. A detailed characterization of KISS1R will help provide link between KISS1 signaling, metabolism and its regulation of dormancy.
Multiple studies have detailed the anti-metastatic role for KP and KISS1R axis; however, the role of full-length KISS1 has not been fully elucidated. Apart from being a precursor molecule for KP, full-length KISS1 interacts with PGC1α-an orchestrator of metabolic event and is enough to suppress metastasis in C8161 melanoma cell line. Therefore, KISS1 processing to KP does not appear to be a requirement to suppress metastasis at least in come cancer types. Furthermore, KISS1 secretion is essential for its anti-metastatic effect, however as discussed earlier KISS1 has a myriad of intracellular functions. Linking the two diverse roles will be required to gain a holistic understanding of its function as a metastasis suppressor.
KP treatments have been employed for progressing through puberty and for alleviating reproductive defects in preclinical models and human patients. However, caution is to be exercised in its administration specially in women due to the reports on its role in breast cancer invasion and metastasis [64]. Similarly, before employing KP to treat metastatic cancers, a thorough consideration on its effects on normal physiological and endocrine activities is important since studies have reported changes in tumor gene signature during puberty [148]. Our preliminary results suggest the existence of new KISS1 fragments that may have an anti-metastatic role (unpublished work). Further research efforts on KISS1, understanding its biochemistry, pharmacokinetics, interaction with tumor microenvironment and a detailed characterization of KISS1R will decide how reliable of a biomarker will the KISS1–KISS1R axis serve in cancer diagnosis as a potential therapeutic drug for treating aggressive metastatic cancers.
References
Padilla SL, Perez JG, Ben-Hamo M et al (2019) Kisspeptin neurons in the arcuate nucleus of the hypothalamus orchestrate circadian rhythms and metabolism. Curr Biol. https://doi.org/10.1016/j.cub.2019.01.022
Qiu J, Rivera HM, Bosch MA et al (2018) Estrogenic-dependent glutamatergic neurotransmission from kisspeptin neurons governs feeding circuits in females. elife. https://doi.org/10.7554/eLife.35656
Herber CB, Krause WC, Wang L et al (2019) Estrogen signaling in arcuate Kiss1 neurons suppresses a sex-dependent female circuit promoting dense strong bones. Nat Commun. https://doi.org/10.1038/s41467-018-08046-4
Guzman S, Brackstone M, Radovick S et al (2018) KISS1/KISS1R in cancer: friend or foe? Front Endocrinol 9(3):437
Franssen D, Tena-Sempere M (2018) The kisspeptin receptor: a key G-protein-coupled receptor in the control of the reproductive axis. Best Pract Res Clin Endocrinol Metab 32:107–123
Bhattacharya M, Babwah AV et al (2015) Kisspeptin: beyond the brain. Endocrinology. https://doi.org/10.1210/en.2014-1915
Ogawa S, Nathan FM, Parhar IS (2014) Habenular kisspeptin modulates fear in the zebrafish. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1314184111
Welch DR, Chen P, Miele ME et al (1994) Microcell-mediated transfer of chromosome 6 into metastatic human C8161 melanoma cells suppresses metastasis but does not inhibit tumorigenicity. Oncogene 9:255–262
Goldberg SF, Miele ME, Hatta N et al (2003) Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res 63:432–440
Lee JH, Miele ME, Hicks DJ et al (1996) KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. https://doi.org/10.1093/jnci/88.23.1731
Harms JF, Welch DR, Miele ME (2003) KISS1 metastasis suppression and emergent pathways. Clin Exp Metastasis 20:11–18
Beck BH, Welch DR (2010) The KISS1 metastasis suppressor: a good night kiss for disseminated cancer cells. Eur J Cancer. https://doi.org/10.1016/j.ejca.2010.02.023
Nash KT, Welch DR (2007) The KISS1 metastasis suppressor: mechanistic insights and clinical utility. Front Biosci. https://doi.org/10.2741/1824
Lee DK, Nguyen T, O’Neill GP et al (1999) Discovery of a receptor related to the galanin receptors. FEBS Lett. https://doi.org/10.1016/S0014-5793(99)00009-5
Kotani M, Detheux M, Vandenbogaerde A et al (2001) The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. https://doi.org/10.1074/jbc.M104847200
Muir AI, Chamberlain L, Elshourbagy NA et al (2001) AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. J Biol Chem. https://doi.org/10.1074/jbc.M102743200
Ohtaki T, Shintani Y, Honda S et al (2001) Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. https://doi.org/10.1038/35079135
Kirby HR, Maguire JJ, Colledge WH, Davenport AP (2010) International union of basic and clinical pharmacology. LXXVII. Kisspeptin receptor nomenclature, distribution, and function. Pharmacol Rev 62:565–578
Takino T, Koshikawa N, Miyamori H et al (2003) Cleavage of metastasis suppressor gene product KiSS-1 protein/metastin by matrix metalloproteinases. Oncogene. https://doi.org/10.1038/sj.onc.1206542
Hook V, Lietz CB, Podvin S et al (2018) Diversity of neuropeptide cell-cell signaling molecules generated by proteolytic processing revealed by neuropeptidomics mass spectrometry. J Am Soc Mass Spectrom. https://doi.org/10.1007/s13361-018-1914-1
Chilumuri A, Milton NGN (2013) The role of neurotransmitters in protection against amyloid-β toxicity by KiSS-1 overexpression in SH-SY5Y neurons. ISRN Neurosci. https://doi.org/10.1155/2013/253210
Milton NGN (2012) In vitro activities of Kissorphin, a novel Hexapeptide KiSS-1 derivative, in neuronal cells. J Amino Acids. https://doi.org/10.1155/2012/691463
Makri A, Pissimissis N, Lembessis P et al (2008) The kisspeptin (KiSS-1)/GPR54 system in cancer biology. Cancer Treat Rev 34:682–692
Shirasaki F, Takata M, Hatta N, Takehara K (2001) Loss of expression of the metastasis suppressor gene KiSS1 during melanoma progression and its association with LOH of chromosome 6q16.3-q23. Cancer Res 61:7422–7425
Stafford LJ, Xia C, Ma W et al (2002) Identification and characterization of mouse metastasis-suppressor KiSS1 and its G-protein-coupled receptor. Cancer Res 62:5399–5404
Seminara SB, Messager S, Chatzidaki EE et al (2004) The GPR54 gene as a regulator of puberty. Obstet Gynecol Surv. https://doi.org/10.1097/00006254-200405000-00020
Carel J-C, Chaussain J-L, Milgrom E et al (2003) Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1834399100
Colledge WH (2009) Transgenic mouse models to study Gpr54/kisspeptin physiology. Peptides. https://doi.org/10.1016/j.peptides.2008.05.006
Dhillo W (2013) Timeline: kisspeptins. Lancet Diabetes Endocrinol 1:12–13
Trevisan CM, Montagna E, De Oliveira R et al (2018) Kisspeptin/GPR54 system: what do we know about its role in human reproduction? Cell Physiol Biochem 49:1259–1276
Silveira LG, Noel SD, Silveira-Neto AP et al (2010) Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab. https://doi.org/10.1210/jc.2009-2421
Topalglu AK, Tello JA, Kotan LD et al (2012) Inactivating KISS1 mutation and hypogonadotropic hypogonadism. Obstet Gynecol Surv. https://doi.org/10.1097/ogx.0b013e31825bc1be
Schmid K, Wang X, Haitel A et al (2007) KiSS-1 overexpression as an independent prognostic marker in hepatocellular carcinoma: an immunohistochemical study. Virchows Arch. https://doi.org/10.1007/s00428-006-0352-9
McNally LR, Welch DR, Beck BH et al (2010) KISS1 over-expression suppresses metastasis of pancreatic adenocarcinoma in a xenograft mouse model. Clin Exp Metastasis. https://doi.org/10.1007/s10585-010-9349-5
Huijbregts L, De Roux N (2010) KISS1 is down-regulated by 17β-estradiol in MDA-MB-231 cells through a nonclassical mechanism and loss of ribonucleic acid polymerase ii binding at the proximal promoter. Endocrinology. https://doi.org/10.1210/en.2010-0260
Teng Y, Mei Y, Hawthorn L, Cowell JK (2014) WASF3 regulates miR-200 inactivation by ZEB1 through suppression of KISS1 leading to increased invasiveness in breast cancer cells. Oncogene. https://doi.org/10.1038/onc.2012.565
Kasum M, Franulić D, Čehić E, Orešković S, Lila A, Ejubović E et al (2014) Kisspeptin: a novel physiological trigger for oocyte maturation in in-vitro fertilisation treatment. Lancet 383:S17
Tena-Sempere M, Felip A, Gómez A et al (2012) Comparative insights of the kisspeptin/kisspeptin receptor system: lessons from non-mammalian vertebrates. Gen Comp Endocrinol 175:234–243
Yeo RL, Tsunekawa K, Mi JM et al (2009) Molecular evolution of multiple forms of kisspeptins and GPR54 receptors in vertebrates. Endocrinology. https://doi.org/10.1210/en.2008-1679
Akazome Y, Kanda S, Okubo K, Oka Y (2010) Functional and evolutionary insights into vertebrate kisspeptin systems from studies of fish brain. J Fish Biol 76:161–182
Beck BH, Fuller SA, Peatman E et al (2012) Chronic exogenous kisspeptin administration accelerates gonadal development in basses of the genus Morone. Comp Biochem Physiol. https://doi.org/10.1016/j.cbpa.2012.03.019
Clarke SA, Dhillo WS (2016) Kisspeptin across the human lifespan:evidence from animal studies and beyond. J Endocrinol. https://doi.org/10.1530/joe-15-0538
Kauffman AS (2010) Coming of age in the kisspeptin era: sex differences, development, and puberty. Mol Cell Endocrinol 324:51–63
Uenoyama Y, Inoue N, Nakamura S, Tsukamura H (2019) Central mechanism controlling pubertal onset in mammals: a triggering role of kisspeptin. Front Endocrinol 61:7422–7425
Tena-Sempere M (2017) Metabolic regulation of reproduction: roles of the hypothalamic Kiss1 system. Biol Reprod. https://doi.org/10.1093/biolreprod/83.s1.197
Jiang Y, Zhu Y, Shi Y, He Y, Kuang Z, Sun Z, Wang J et al (2013) Downregulation of SPARC expression inhibits the invasion of human trophoblast cells in vitro. PLoS ONE. https://doi.org/10.1371/journal.pone.0069079
Babwah AV (2015) Uterine and placental KISS1 regulate pregnancy: what we know and the challenges that lie ahead. Reproduction 150:R121–R128
Tena-Sempere M (2014) Physiological mechanisms for the metabolic control of reproduction. In: Knobil E, Neill JD (eds) Physiology of reproduction: two-volume set. Academic Press, London
Tolson KP, Marooki N, Wolfe A et al (2019) Cre/lox generation of a novel whole-body Kiss1r KO mouse line recapitulates a hypogonadal, obese, and metabolically-impaired phenotype. Mol Cell Endocrinol 498:110559. https://doi.org/10.1016/j.mce.2019.110559
Song WJ, Mondal P, Wolfe A et al (2014) Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab. https://doi.org/10.1016/j.cmet.2014.03.005
Welch DR, Hurst DR (2019) Defining the hallmarks of metastasis. Cancer Res 79:3011–3027
Stafford LJ, Vaidya KS, Welch DR (2008) Metastasis suppressors genes in cancer. Int J Biochem Cell Biol 40:843–847
Hurst DR, Welch DR (2011) Metastasis suppressor genes. At the interface between the environment and tumor cell growth. Int Rev Cell Mol Biol 286:107–180
Smith SC, Theodorescu D (2009) Learning therapeutic lessons from metastasis suppressor proteins. Nat Rev Cancer 9:253–264
Eccles SA, Welch DR (2007) Metastasis: recent discoveries and novel treatment strategies. Lancet. https://doi.org/10.1016/S0140-6736(07)60781-8
West A, Vojta PJ, Welch DR, Weissman BE (1998) Chromosome localization and genomic structure of the KiSS-1 metastasis suppressor gene (KISS1). Genomics. https://doi.org/10.1006/geno.1998.5566
Shin R, Welch DR, Mishra VK et al (2009) Nuclear magnetic resonance and circular dichroism study of metastin (Kisspeptin-54) structure in solution. Clin Exp Metastasis. https://doi.org/10.1007/s10585-009-9252-0
Swain SS, Mohanty S, Panda D et al (2012) In silico structural analysis and characterization of human Kiss-1 receptor: a metastasis suppressor protein in melanomas and breast cancer. J Endocrinol Reprod 52:3
De Opakua AI, Merino N, Villate M et al (2017) The metastasis suppressor KISS1 is an intrinsically disordered protein slightly more extended than a random coil. PLoS ONE. https://doi.org/10.1371/journal.pone.0172507
Nash KT, Phadke PA, Navenot JM et al (2007) Requirement of KISS1 secretion for multiple organ metastasis suppression and maintenance of tumor dormancy. J Natl Cancer Inst. https://doi.org/10.1093/jnci/djk053
Harihar S, Pounds KM, Iwakuma T et al (2014) Furin is the major proprotein convertase required for KISS1-to-Kisspeptin processing. PLoS ONE. https://doi.org/10.1371/journal.pone.0084958
Zheng S, Chang Y, Hodges KB et al (2010) Expression of KISS1 and MMP-9 in non-small cell lung cancer and their relations to metastasis and survival. Anticancer Res 30:713–718
Jiang Y, Berk M, Singh LS et al (2005) KiSS1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase C alpha. Clin Exp Metastasis. https://doi.org/10.1007/s10585-005-8186-4
Cvetković D, Babwah AV, Bhattacharya M (2013) Kisspeptin/KISSIR system in breast cancer. J Cancer 4(8):653–661
Chen Y, Zhang C, Chen J et al (2018) Expression of transcription factor 21 (TCF21) and upregulation its level inhibits invasion and metastasis in esophageal squamous cell carcinoma. Med Sci Monit. https://doi.org/10.12659/msm.909138
Cebrian V, Fierro M, Orenes-Piero E et al (2011) KISS1 methylation and expression as tumor stratification biomarkers and clinical outcome prognosticators for bladder cancer patients. Am J Pathol. https://doi.org/10.1016/j.ajpath.2011.05.009
Schmidt E, Haase M, Ziegler E et al (2014) Kisspeptin-10 inhibits stromal-derived factor 1-induced invasion of human endometrial cancer cells. Int J Gynecol Cancer. https://doi.org/10.1097/IGC.0000000000000050
Zhang Y, Tang YJ, Li ZH et al (2013) KiSS1 inhibits growth and invasion of osteosarcoma cells through inhibition of the MAPK pathway. Eur J Histochem. https://doi.org/10.4081/ejh.2013.e30
Bin SY, Xu S (2013) Expression of KISS1 and KISS1R (GPR54) may be used as favorable prognostic markers for patients with non-small cell lung cancer. Int J Oncol. https://doi.org/10.3892/ijo.2013.1967
Okugawa Y, Inoue Y, Tanaka K et al (2013) Loss of the metastasis suppressor gene KiSS1 is associated with lymph node metastasis and poor prognosis in human colorectal cancer. Oncol Rep. https://doi.org/10.3892/or.2013.2558
Martins CMO, Fernandes BF, Antecka E et al (2008) Expression of the metastasis suppressor gene KISS1 in uveal melanoma. Eye. https://doi.org/10.1038/sj.eye.6703090
Lee JH, Welch DR (1997) Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res 57:2384–2387
Kostakis ID, Agrogiannis G, Vaiopoulos AG et al (2015) A clinicopathological analysis of KISS1 and KISS1R expression in colorectal cancer. APMIS. https://doi.org/10.1111/apm.12397
Kostakis ID, Agrogiannis G, Vaiopoulos AG et al (2018) KISS1 and KISS1R expression in gastric cancer. J BUON 23:79–84
Jiffar T, Yilmaz T, Lee J et al (2011) KiSS1 mediates platinum sensitivity and metastasis suppression in head and neck squamous cell carcinoma. Oncogene. https://doi.org/10.1038/onc.2011.39
Cho SG, Wang Y, Rodriguez M et al (2011) Haploinsufficiency in the prometastasis Kiss1 receptor Gpr54 delays breast tumor initiation, progression, and lung metastasis. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-11-0329
Ringel MD, Hardy E, Bernet VJ et al (2002) Metastin receptor is overexpressed in papillary thyroid cancer and activates MAP kinase in thyroid cancer cells. J Clin Endocrinol Metab. https://doi.org/10.1210/jc.87.5.2399
Platonov ME, Borovjagin AV, Kaverina N, Xiao T, Kadagidze Z, Lesniak M, Baryshnikova M, Ulasov IV et al (2018) KISS1 tumor suppressor restricts angiogenesis of breast cancer brain metastases and sensitizes them to oncolytic virotherapy in vitro. Cancer Lett. https://doi.org/10.1016/j.canlet.2017.12.024
Stallaert W, Brüggemann Y, Sabet O et al (2018) Contact inhibitory Eph signaling suppresses EGF-promoted cell migration by decoupling EGFR activity from vesicular recycling. Sci Signal. https://doi.org/10.1126/scisignal.aat0114
Kim TH, Cho SG (2017) Melatonin-induced KiSS1 expression inhibits triple-negative breast cancer cell invasiveness. Oncol Lett. https://doi.org/10.3892/ol.2017.6434
Goertzen CG, Dragan M, Turley E et al (2016) KISS1R signaling promotes invadopodia formation in human breast cancer cell via β-arrestin2/ERK. Cell Signal. https://doi.org/10.1016/j.cellsig.2015.12.010
Zajac M, Law J, Cvetkovic DD et al (2011) GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS ONE. https://doi.org/10.1371/journal.pone.0021599
Cvetković D, Dragan M, Leith SJ et al (2013) KISS1R induces invasiveness of estrogen receptor-negative human mammary epithelial and breast cancer cells. Endocrinology. https://doi.org/10.1210/en.2012-2164
Marot D, Bieche I, Aumas C et al (2007) High tumoral levels of Kiss1 and G-protein-coupled receptor 54 expression are correlated with poor prognosis of estrogen receptor-positive breast tumors. Endocr Relat Cancer. https://doi.org/10.1677/ERC-07-0012
Blake A, Dragan M, Tirona RG et al (2017) G protein-coupled KISS1 receptor is overexpressed in triple negative breast cancer and promotes drug resistance. Sci Rep. https://doi.org/10.1038/srep46525
Tian J, Al-Odaini AA, Wang Y et al (2018) KiSS1 gene as a novel mediator of TGFβ-mediated cell invasion in triple negative breast cancer. Cell Signal. https://doi.org/10.1016/j.cellsig.2017.10.002
Ikeguchi M, Hirooka Y, Kaibara N (2003) Quantitative reverse transcriptase polymerase chain reaction analysis for KiSS-1 and orphan G-protein-coupled receptor (hOT7T175) gene expression in hepatocellular carcinoma. J Cancer Res Clin Oncol. https://doi.org/10.1007/s00432-003-0469-z
Zang S, Liu JF, Wang B et al (2009) Expression of KiSS-1 gene and its role in invasion and metastasis of human hepatocellular carcinoma. Anat Rec. https://doi.org/10.1002/ar.20950
Song WW, Gui AP, Li W et al (2017) Expressions of HIF-1α and KISS-1 in patients with liver cancer and correlation analysis. Eur Rev Med Pharmacol Sci 21(18):4058–4063
Savvidis C, Papaoiconomou E, Petraki C, Msaouel P, Koutsilieris M et al (2015) The role of KISS1/KISS1R system in tumor growth and invasion of differentiated thyroid cancer. Anticancer Res 35(2):819–826
Jabeen S, Zahid Qureshi M, Javed Z et al (2016) Kisspeptin mediated signaling in cancer. Curr Top Med Chem. https://doi.org/10.2174/1568026616666160212123309
Kaverina N, Borovjagin AV, Kadagidze Z et al (2017) Astrocytes promote progression of breast cancer metastases to the brain via a KISS1-mediated autophagy. Autophagy. https://doi.org/10.1080/15548627.2017.1360466
Sun W, Li S, Yu Y et al (2019) MicroRNA-3648 Is upregulated to suppress TCF21, resulting in promotion of invasion and metastasis of human bladder cancer. Mol Ther Nucleic Acids. https://doi.org/10.1016/j.omtn.2019.04.006
Zhang H, Guo Y, Shang C et al (2012) MiR-21 downregulated TCF21 to inhibit KISS1 in renal cancer. Urology. https://doi.org/10.1016/j.urology.2012.08.013
Arab K, Smith LT, Gast A et al (2011) Epigenetic deregulation of TCF21 inhibits metastasis suppressor KISS1 in metastatic melanoma. Carcinogenesis. https://doi.org/10.1093/carcin/bgr138
Teng Y, Bahassan A, Dong D et al (2016) Targeting the WASF3-CYFIP1 complex using stapled peptides suppresses cancer cell invasion. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-15-1680
Teng Y, Liu M, Cowell JK (2011) Functional interrelationship between the WASF3 and KISS1 metastasis-associated genes in breast cancer cells. Int J Cancer. https://doi.org/10.1002/ijc.25964
Shen Z, Wang B, Jiang K et al (2016) Downregulation of miR-199b is associated with distant metastasis in colorectal cancer via activation of SIRT1 and inhibition of CREB/KISS1 signaling. Oncotarget. https://doi.org/10.18632/oncotarget.9042
Liu G, Zhao X, Zhou J et al (2018) LncRNA TP73-AS1 promotes cell proliferation and inhibits cell apoptosis in clear cell renal cell carcinoma through repressing KISS1 expression and inactivation of PI3K/Akt/mTOR signaling pathway. Cell Physiol Biochem. https://doi.org/10.1159/000491767
Qiu J, Lin X, Tang X et al (2019) Long noncoding RNA TC0101441 induces epithelial-mesenchymal transition in epithelial ovarian cancer metastasis by downregulating KiSS1. Int J Cancer. https://doi.org/10.1002/ijc.32692
Zhang YX, Cui HX, Liu L, Yi GK (2019) Long non-coding RNA MNX1-AS1 promoted osteosarcoma proliferation and invasion via inhibiting KISS1. Eur Rev Med Pharmacol Sci. https://doi.org/10.26355/eurrev_201907_18417
Liu C, Wang L, Li YW, Cui YS, Wang YQLS (2019) Long noncoding RNA LUCAT1 promotes migration and invasion of prostate cancer cells by inhibiting KISS1 expression. Eur Rev Med Pharmacol Sci 23:3277–3283
Mitra A, Fillmore RA, Metge BJ et al (2008) Large isoform of MRJ (DNAJB6) reduces malignant activity of breast cancer. Breast Cancer Res. https://doi.org/10.1186/bcr1874
Zhang Y, Huang Z, Zhu Z, Zheng X, Liu J, Han Z, Ma X, Zhang Y et al (2014) Upregulated UHRF1 promotes bladder cancer cell invasion by epigenetic silencing of KiSS1. PLoS ONE. https://doi.org/10.1371/journal.pone.0104252
Alhosin M, Omran Z, Zamzami MA et al (2016) Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. J Exp Clin Cancer Res 35:174
Babwah AV, Pampillo M, Min L, Kaiser UB, Bhattacharya M (2012) Cells undergo sustained kisspeptin-induced signaling that is dependent upon an influx of extracellular Ca2+. Endocrinology. https://doi.org/10.1210/en.2012-1747
Martínez-Fuentes AJ, Molina M, Vázquez-Martínez R et al (2011) Expression of functional KISS1 and KISS1R system is altered in human pituitary adenomas: Evidence for apoptotic action of kisspeptin-10. Eur J Endocrinol. https://doi.org/10.1530/EJE-10-0905
Esposito S, Russo MV, Airoldi I et al (2015) SNAI2/Slug gene is silenced in prostate cancer and regulates neuroendocrine differentiation, metastasis-suppressor and pluripotency gene expression. Oncotarget. https://doi.org/10.18632/oncotarget.2736
Tan K, Cho S-G, Luo W et al (2014) KiSS1-induced GPR54 signaling inhibits breast cancer cell migration and epithelial-mesenchymal transition via protein kinase D1. Curr Mol Med. https://doi.org/10.2174/1566524014666140603115314
Cho SG, Li D, Stafford LJ et al (2009) KiSS1 suppresses TNFα-induced breast cancer cell invasion via an inhibition of RhoA-mediated NF-κB activation. J Cell Biochem. https://doi.org/10.1002/jcb.22216
Song GQ, Zhao Y (2015) Kisspeptin-10 inhibits the migration of breast cancer cells by regulating epithelial-mesenchymal transition. Oncol Rep. https://doi.org/10.3892/or.2014.3619
Navenot J-M, Fujii N, Peiper SC (2009) Activation of rho and rho-associated kinase by GPR54 and KiSS1 metastasis suppressor gene product induces changes of cell morphology and contributes to apoptosis. Mol Pharmacol. https://doi.org/10.1124/mol.109.055095
Cho SG, Yi Z, Pang X et al (2009) Kisspeptin-10, a KISS1-derived decapeptide, inhibits tumor angiogenesis by suppressing Sp1-mediated VEGF expression and FAK/Rho GTPase activation. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-09-0476
Deng G, Zheng X, Jiang P et al (2017) Notch1 suppresses prostate cancer cell invasion via the metastasis-associated 1-KiSS-1 metastasis-suppressor pathway. Oncol Lett. https://doi.org/10.3892/ol.2017.6761
Kauffman EC, Robinson VL, Stadler WM et al (2003) Metastasis suppression: the evolving role of metastasis suppressor genes for regulating cancer cell growth at the secondary site. J Urol 169:1122–1133
Kim TH, Cho SG (2017) Kisspeptin inhibits cancer growth and metastasis via activation of EIF2AK2. Mol Med Rep. https://doi.org/10.3892/mmr.2017.7578
Navenot JM, Wang Z, Chopin M et al (2005) Kisspeptin-10-induced signaling of GPR54 negatively regulates chemotactic responses mediated by CXCR4: a potential mechanism for the metastasis suppressor activity of kisspeptins. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-05-1757
Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7:834–846
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14:611–622
Linde N, Fluegen G, Aguirre-Ghiso JA (2016) The relationship between dormant cancer cells and their microenvironment. Adv Cancer Res 132:45–71
Klein CA (2011) Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev 21:42–49
Vallette FM, Olivier C, Lézot F et al (2019) Dormant, quiescent, tolerant and persister cells: four synonyms for the same target in cancer. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2018.11.004
Horak CE, Lee JH, Marshall JC et al (2008) The role of metastasis suppressor genes in metastatic dormancy. APMIS 116:586–601
Goldberg SF, Harms JF, Quon K, Welch DR (1999) Metastasis-suppressed C8161 melanoma cells arrest in lung but fail to proliferate. Clin Exp Metastasis. https://doi.org/10.1023/A:1006718800891
Corno C, Perego P (2019) KiSS1 in regulation of metastasis and response to antitumor drugs. Drug Resist Updat 42:12–21
Lee KH, Kim JR (2009) Kiss-1 suppresses MMP-9 expression by activating p38 MAP kinase in human stomach cancer. Oncol Res. https://doi.org/10.3727/096504009789954591
Navenot J-M, Fujii N, Peiper SC (2009) KiSS1 metastasis suppressor gene product induces suppression of tyrosine kinase receptor signaling to Akt, tumor necrosis factor family ligand expression, and apoptosis. Mol Pharmacol. https://doi.org/10.1124/mol.108.054270
Gonzalez CD, Alvarez S, Ropolo A et al (2014) Autophagy, warburg, and warburg reverse effects in human cancer. Biomed Res Int. https://doi.org/10.1155/2014/926729
Ulasov IV, Borovjagin AV, Timashev P et al (2019) KISS1 in breast cancer progression and autophagy. Cancer Metastasis Rev 38:493–506. https://doi.org/10.1007/s10555-019-09814-4
Wang CH, Qiao C, Wang RC, Zhou WP (2016) KiSS-1-mediated suppression of the invasive ability of human pancreatic carcinoma cells is not dependent on the level of KiSS-1 receptor GPR54. Mol Med Rep. https://doi.org/10.3892/mmr.2015.4535
Young ED, Strom K, Tsue AF et al (2018) Automated quantitative image analysis for ex vivo metastasis assays reveals differing lung composition requirements for metastasis suppression by KISS1. Clin Exp Metastasis. https://doi.org/10.1007/s10585-018-9882-1
Smith HA, Kang Y (2013) The metastasis-promoting roles of tumor-associated immune cells. J Mol Med 91:411–429
Kitamura T, Qian BZ, Pollard JW (2015) Immune cell promotion of metastasis. Nat Rev Immunol 15:73–86
Lu J, Tan M, Cai Q (2015) The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 356:156–164
Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell. https://doi.org/10.1016/j.cell.2008.08.021
Cantor JR, Sabatini DM (2012) Cancer cell metabolism: one hallmark, many faces. Cancer Discov 2:881–898
Liu W, Beck BH, Vaidya KS et al (2014) Metastasis suppressor KISS1Seemsto reverse the warburg effect by enhancing mitochondrial biogenesis. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-13-1183
Manley SJ, Liu W, Welch DR (2017) The KISS1 metastasis suppressor appears to reverse the Warburg effect by shifting from glycolysis to mitochondrial beta-oxidation. J Mol Med. https://doi.org/10.1007/s00109-017-1552-2
Squillacciotti S, Krämer K, Füssel S, Meinhardt M (2011) Diagnostic value of plasmatic Kp-10 and prognostic relevance of kisspeptin and KISS1R expression in clear cell renal cell carcinoma (ccRCC). Eur Urol Suppl 10(2):106
Zuco V, Cassinelli G, Cossa G et al (2015) Targeting the invasive phenotype of cisplatin-resistant Non-Small Cell Lung Cancer cells by a novel histone deacetylase inhibitor. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2015.01.002
Tanaka A, Nakata D, Masaki T et al (2018) Evaluation of pharmacokinetics/pharmacodynamics and efficacy of one-month depots of TAK-448 and TAK-683, investigational kisspeptin analogs, in male rats and an androgen-dependent prostate cancer model. Eur J Pharmacol 822:138–146. https://doi.org/10.1016/j.ejphar.2018.01.012
Ciaramella V, Della Corte CM, Ciardiello F, Morgillo F (2018) Kisspeptin and cancer: Molecular interaction, biological functions, and future perspectives. Front Endocrinol (Lausanne) 9:115
Ciaramella V, Della Corte CM, Di Mauro C et al (2018) Antitumor efficacy of Kisspeptin in human malignant mesothelioma cells. Oncotarget. https://doi.org/10.18632/oncotarget.25018
Dhillo WS, Chaudhri OB, Patterson M et al (2005) Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab. https://doi.org/10.1210/jc.2005-1468
Jayasena CN, Comninos AN, Veldhuis JD et al (2013) A single injection of kisspeptin-54 temporarily increases luteinizing hormone pulsatility in healthy women. Clin Endocrinol. https://doi.org/10.1111/cen.12179
Jayasena CN, Nijher GMK, Comninos AN et al (2011) The effects of kisspeptin-10 on reproductive hormone release show sexual dimorphism in humans. J Clin Endocrinol Metab. https://doi.org/10.1210/jc.2011-1408
De Tassigny XDA, Jayasena C, Murphy KG et al (2017) Mechanistic insights into the more potent effect of KP-54 compared to KP-10 in vivo. PLoS ONE. https://doi.org/10.1371/journal.pone.0176821
Roth CL, Mastronardi C, Lomniczi A et al (2007) Expression of a tumor-related gene network increases in the mammalian hypothalamus at the time of female puberty. Endocrinology. https://doi.org/10.1210/en.2007-0634
Acknowledgements
The authors would like to thank Keerthika J. Jayachandran for help with graphical design for Fig. 1. The authors would like to thank past and present members of Harihar and Welch Laboratories for helpful discussions and suggestions. The authors appreciate direct financial support from selective excellence initiative grant, SRM Institute of Science and Technology (SH), Susan G. Komen for the Cure SAC110037 (DRW), National Foundation for Cancer Research (DRW) and P30-CA168524 (DRW).
Author information
Authors and Affiliations
Contributions
SH and DRW had the idea for the article. SH, SR, SN, AS, TY and DRW performed the literature search. SH and DRW drafted and critically revised the work. All authors approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Harihar, S., Ray, S., Narayanan, S. et al. Role of the tumor microenvironment in regulating the anti-metastatic effect of KISS1. Clin Exp Metastasis 37, 209–223 (2020). https://doi.org/10.1007/s10585-020-10030-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-020-10030-6