
Abstract
Ovarian cancer, the most lethal gynaecological cancer, is characterised by the shedding of epithelial cells from the ovarian surface, followed by metastasis and implantation onto the peritoneal surfaces of abdominal organs. Our proteomic studies investigating the interactions between peritoneal (LP-9) and ovarian cancer (OVCAR-5) cells found transketolase (TKT) to be regulated in the co-culture system. This study characterized TKT expression in advanced stage (III/IV) serous ovarian cancers (n = 125 primary and n = 54 peritoneal metastases), normal ovaries (n = 6) and benign serous cystadenomas (n = 10) by immunohistochemistry. In addition, we also evaluated the function of TKT in ovarian cancer cells in vitro. Nuclear TKT was present in all primary serous ovarian cancer tissues examined (median 82.0 %, range 16.5–100 %) and was significantly increased in peritoneal metastases compared with matching primary cancers (P = 0.01, Wilcoxon Rank test). Kaplan–Meier survival and Cox regression analyses showed that high nuclear TKT positivity in peritoneal metastases (>94 %) was significantly associated with reduced overall survival (P = 0.006) and a 2.8 fold increased risk of ovarian cancer death (95 % CI 1.29–5.90, P = 0.009). Knockdown of TKT by siRNAs significantly reduced SKOV-3 cell proliferation but had no effect on their motility or invasion. Oxythiamine, an inhibitor of TKT activity, significantly inhibited the proliferation of four ovarian cancer cell lines (OV-90, SKOV-3, OVCAR-3 and OVCAR-5) and primary serous ovarian cancer cells isolated from patient ascites. In conclusion, these findings indicate that TKT plays an important role in the proliferation of metastatic ovarian cancer cells and could be used as novel therapeutic target for advanced disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ovarian cancer is the most lethal gynaecological cancer and the fifth most common cause of cancer related death among Western women [1]. Although ovarian cancers represent only 30 % of cancers of the female genital tract, they are responsible for half of the deaths [1]. The disproportionately high mortality rate from ovarian cancer is attributed to the late presentation of the disease (stage III or IV). Survival rate of ovarian cancer women has not improved considerably over the last 20 years, highlighting the urgency for developing new diagnostic methods and more effective therapeutics against this disease.
Almost 85 % of ovarian cancers are thought to originate at the ovarian surface epithelium [2]. The cells that undergo neoplastic transformations are shed and disseminate into the peritoneal cavity. They adhere to the peritoneum lining, proliferate, migrate, and invade the abdominal organs forming secondary metastatic sites. The peritoneal microenvironment is believed to be an active participant in the process of ovarian cancer metastasis. Implantation and invasion occurs within a tumor–host interface where cancer and peritoneal cells exchange proteins and peptides, which modify the local extracellular environment, stimulate migration, and promote proliferation and invasion [3–9]. A greater understanding of these processes would potentially lead to the discovery of novel molecular targets to block this critical step of ovarian cancer metastasis. Our group has recently explored the interactions between ovarian cancer-peritoneal cell interactions, using an in vitro co-culture system with a proteomic approach [10, 11]. One of the proteins found to be regulated by ovarian cancer–peritoneal cell interactions was transketolase (TKT).
TKT is a metabolic enzyme that catalyses key reactions in the non-oxidative branch of pentose phosphate pathway (PPP) [12]. TKT exists as a homodimer consisting of two identical 75 kDa subunits with the active site at their interface between the contacting monomers [12, 13]. TKT requires the binding of one molecule of thiamine diphosphate (TDP) and a bivalent cation like Ca2+ or Mg2+ for its activation [12]. TKT catalyses reactions which: (1) reversibly convert pentose phosphates to intermediate six and three carbon sugars for further use in the glycolysis pathway and (2) generate de novo ribose-5-phosphate, which is necessary for the synthesis of nucleic acids [14]. Due to its important role in glucose metabolism, reduced levels of TKT due to thiamine deficiency or genetic defects have been associated with various conditions such as Wernicke’s encephalopathy, Alzheimer’s disease and cancer [15–17].
The TKT pathway has been shown to produce >70 % of ribose necessary for nucleotide synthesis in various cancers such as those of the pancreas, liver and lung, and therefore is thought to play a major role in the proliferation of cancer cells which require increased nucleotide synthesis for their growth [17–19]. Inhibiting TKT activity using irreversible inhibitors such as oxythiamine (an inactive analogue of TKT co-factor thiamine) was shown to inhibit in vitro pancreatic cancer cell proliferation by 39 % and Ehrlich tumor growth in vivo by 90 % [17, 20]. Oxythiamine treatment was non-toxic and inhibited Ehrlich’s tumor cell growth by arresting cells in the G1 phase of cell cycle [20]. Together these studies have provided strong evidence that TKT plays an important role in tumor growth. However, to date no studies have characterised its expression and potential role in ovarian cancer. We have therefore characterised the expression of TKT in a range of commercial ovarian cancer cell lines, primary serous carcinoma cells as well as serous ovarian cancer tissues and determined its role in the proliferation and metastatic behaviour of serous ovarian cancer cells. To our knowledge, this is the first study to investigate the function of TKT in ovarian cancer.
Materials and methods
Cell culture
The human ovarian cancer cell lines OVCAR-3, SKOV-3, and OV-90 were purchased from American Type Culture Collection (ATCC, VA, USA). OVCAR-5 cells were obtained from Dr Thomas Hamilton (Fox Chase Cancer Center, PA, USA) and peritoneal LP-9 cells were purchased from Coriell Cell Repositories (Camden, NJ, USA). All cell lines were maintained at 37 °C in an environment of 5 % CO2 as previously described [10].
Primary ovarian cancer cells were derived from ascites collected from advanced stage serous ovarian cancer patients with informed patient consent and the approval from the Royal Adelaide Hospital Human Ethics Committee. Ascites (~50 ml) was centrifuged at room temperature at 3000 rpm for 10 min and the cell pellet was resuspended in media and maintained in SensiCell™ RPMI 1640 (Gibco) medium supplemented with 4 mM l-glutamine, 10 % FBS (Sigma Aldrich, St Louis, MO, USA) and antibiotics (100 U penicillin G, 100 µg/ml streptomycin sulfate and 100 µg/ml amphotericin B (Sigma Aldrich). Primary ovarian cancer cell lines were stored in liquid nitrogen at P2 and were used in subsequent experiments between P3 and P5. The clinico-pathological characteristics of the patients whose ascites was used to isolate the primary cells are shown in Supplementary Table 1.
Direct co-culture of ovarian cancer and peritoneal cells
Peritoneal LP-9 cells were cultured in 6-well plates in their growth medium until they reached confluency. The monolayer was then washed with and incubated in serum free LP-9 media for 1.5 h at 37 °C at 5 % CO2. OVCAR-5 and SKOV-3 cells were washed separately, following the same procedure as for LP-9 monolayer, trypsinized and centrifuged at 1500 rpm for 5 min. They were then washed, resuspended in serum-free LP-9 media, added (at 2 × 105 cells/well concentration) to the LP-9 monolayer and co-cultured for 48 h at 37 °C and 5 % CO2 before collection of the conditioned media (CM) as previously described [10]. OVCAR-5 and LP-9 cells were also co-cultured in the presence or absence α2-antiplasmin (0.2 µM, Calbiochem, Merck Millipore, Darmstadt, Germany). CM was precipitated with 4 volumes of 100 % cold acetone at −20 °C overnight.
2D electrophoresis
Immobilized pH gradient (IPG) strips (11 cm, GE Healthcare, Little Chalfont, England) were rehydrated overnight in 200 µl rehydration buffer (1.2 % (v/v) DeStreak, 0.5 % pH 3–11 IPG buffer, 7 M urea, 2 M thiourea, 4 % CHAPS, 30 mM Tris and a trace amount of bromophenol blue). Protein samples (50 µg) from the precipitated CM of a mix of LP-9 and OVCAR-5 single cultures, and co-cultured LP-9 and OVCAR-5 were dissolved in lysis buffer containing 7 M urea, 2 M thiourea, 4 % CHAPS, 0.8 % IPG buffer, 65 mM DTT, 1 mM PMSF, and 30 mM Tris and loaded onto the rehydrated IPG strips. Isoelectric focusing was carried out and proteins were separated at room temperature using a step-wise gradient until 46,000 Vhrs was achieved using the Ettan IPGphor II system (GE Healthcare). Following equilibration, the IPG strips were applied to the Criterion XT precast gels 4-12 % Bis–Tris (Bio-Rad, Hercules, USA). SDS polyacrylamide gel electrophoresis (SDS-PAGE) was performed at 50 V for 0.5 h and then 150 V until the dye-front reached the bottom. Gels containing protein from LP-9 and OVCAR-5 cultured alone and LP-9 and OVCAR-5 co-cultured cells were stained with silver stain (Biorad), scanned, and analysed using PDQuest advanced 2D analysis software (v 7.0, Biorad). Gels were warped using identified landmark spots present in every gel as anchor points. Warped gels were overlaid in pairs and viewed in darkview in the multichannel viewer to determine which spots were differentially expressed in the LP-9 and OVCAR-5 co-cultured CM media when compared with the mix of LP-9 and OVCAR-5 proteins cultured alone. All experiments were performed in duplicate and only spots identified as differentially expressed in both experiments were selected for mass spectrometry identification.
Mass spectrometry
Differentially expressed protein spots were excised from the gels, destained, and digested with 50 ng of trypsin per sample. A control gel piece was cut from a blank portion of a gel as a negative control. The samples (5 µl from ~10 µl) were chromatographed using an Agilent Protein ID Chip column assembly (40 nl trap column with 0·075 × 43 mm C-18 analytical column) housed in an Agilent HPLC-Chip Cube Interface connected to a HCT ultra 3D-Ion-Trap mass spectrometer (Bruker Daltonik, Bremen, Germany). The column was equilibrated with 4 % acetonitrile (ACN)/0.1 % FA at 0.5 μl/min and the samples eluted with an ACN gradient (4–31 % in 32 min). Ionizable species (300 < m/z < 1200) were trapped and two of the most intense ions eluting at the time were fragmented by collision-induced dissociation. MS and MS/MS spectra were subjected to peak detection using DataAnalysis (Version 3.4, Bruker Daltonik) then imported into BioTools (version 3.1, Bruker Daltonik). The MS/MS spectra were submitted to the in-house MASCOT database-searching engine (version 2.2, Matrix Science). Using the following specifications: Taxonomy: mammals, Database: Swissprot 56.7, Enzyme: Trypsin, Fixed modifications: Carbamidomethyl (C), Variable modifications: Oxidation (M), Mass tol MS: 0.3 Da, MS/MS tol: 0.4 Da, Peptide charge: 1+, 2+ and 3+, Missed cleavages: 1.
Quantitative real-time PCR
Ovarian cancer cells were plated at 5,000 cells/well in 96 well plates and cultured until confluency for 72–96 h. Total RNA was isolated and reversely transcribed using the TaqMan® Gene expression Cells-to-CT™ kit (Applied Biosystems, Life Tecnhologies Australia, Mulgrave, Australia), as per the manufacturer’s instructions. Briefly, lysis solution with DNAse was added to each well and incubated for 5 min at room temperature. Stop solution was then added to each well and mixed. The lysate (10 µl) was added to a 40 µl reverse transcription master mix and reverse transcribed for 1 h. Resultant cDNA was stored in 50 µl aliquots at −20 °C for qRT-PCR analysis. qRT-PCR reactions were performed using the Quantstudio 12 K Flex Real Time PCR System (Applied Biosystems). Briefly, PCR reactions were made up to 10 µl and contained TaqMan® Gene Expression Master Mix (2X), TKT primers (HS01115545-m1, span exon 3 and exon 4 of TKT gene, 60 bp amplicon), nuclease free water, and the cDNA. PCR cycling conditions were as follows: 50 °C for 2 min, 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. CT values were normalised to the house keeping gene β-actin and calibrated to OVCAR-3 expression using the 2−∆∆CT method.
Western immunoblotting
OVCAR-5, OVCAR-3, OV-90, SKOV-3, and LP-9 cells were grown to 80 % confluency in 75 cm2 flasks and cell extracts were collected. Cells were dislodged using a cell scraper and resuspended in 200 μl of RIPA buffer (1 % Nonidet P-40, 1 % sodium deoxycholate, 0.1 % SDS, 0.15 M sodium chloride, 50 mM Tris- HCl and 1 mM EDTA, pH 8.0 with protease inhibitor) and spun at 7000 rpm for 10 min. Bradford protein assay (Biorad, Hercules, CA, USA) was used to determine the protein concentration in cell extracts, prior to storage at −20 °C. Equal amounts of protein from all cell extracts were diluted in 5× loading buffer (50 % glycerol, 250 mM Tris pH 6.8, 10 % SDS, 0.5 % bromophenol blue). Recombinant TKT (1 µg) (Abnova, H0007086, Taipei, Taiwan) was used as a positive control. The samples containing 0.05 % v/v β-mercaptoethanol were then heated at 96 °C for 5 min before loading onto 10 % acrylamide gels (Bio-Rad, CA), electrophoresed at a constant 100 V in Tris–Glycine running buffer (Invitrogen) and subsequently transferred to PVDF membranes (GE Healthcare) overnight at 4 °C and a constant 33 V. Non specific binding was blocked with 3 % skim milk powder in TBS containing 0.1 % Tween 20 (Sigma-Aldrich) for 30 min followed by incubation with rabbit polyclonal TKT antibodies 11039-1-AP (1/1000, Proteintech Group, Inc, Chicago, IL, USA) or H-50 (1/100, Santa Cruz Biotechnology, CA, USA) for 2 h. Protein bands on membranes were then incubated with anti-rabbit IgG peroxidase-conjugated secondary antibodies (1/2000, Dako Australia, Sydney, Australia) for 1 h and protein bands were visualized by enhanced chemiluminescence and autoradiography (ECL Hyperfilm, GE Healthcare).
Cytokeratin immunocytochemistry
The epithelial nature of the primary ovarian cancer cells was confirmed by cytokeratin immunocytostaining. Briefly, primary ovarian cancer cells were plated (5 × 104 cells/well) in 8-well tissue culture chamber slides (Nunclon™ Lab-Tek II Chamber slide, RS Glass Slide, Naperville, IL, USA) overnight, maintained at 37 °C in an environment of 5 % CO2. Cells were subsequently fixed with methanol (−20 °C) followed by acetone (−20 °C) and PBS washes. The endogenous peroxidase activity was quenched with 0.3 % H2O2 and slides were blocked with 5 % goat serum (Sigma Aldrich) for 30 min prior to incubating with monoclonal mouse anti-human cytokeratin clone (AE1/AE3, 1/100, Dako Australia). Cytokeratin immunoreactivity was detected sequentially with a biotinylated goat anti-mouse (1/400, Dako Australia), followed by streptavidin-HRP conjugate (1/500, Dako Australia) and diaminobenzidine tetrahydrochloride (Sigma-Aldrich). The slides were counterstained with haematoxylin (Sigma-Aldrich), dehydrated and mounted in Pertex (Medite Medizintechnik, Burgdorf, Germany). Examples of the cytokeratin immunostaining in the primary cells are shown in Supplementary Fig. 1.
TKT immunohistochemistry
Tissue sections from normal ovaries (n = 6), benign serous tumors (n = 10), 174 serous ovarian cancers (125 primary tumors and 54 omental metastases) were analysed by immunohistochemistry. The ovarian cancer tissues were assembled as tissue microarrays from archived specimens obtained from patients treated for FIGO stage III/IV disease at the Royal Adelaide Hospital, South Australia between 1988 and 2011 with the approval of the Royal Adelaide Hospital ethics committee. Detailed pathological and clinical characteristics of the patient cohort are shown in Table 1. Immunohistochemistry was performed on tissue sections (4 μm) using a rabbit polyclonal TKT antibody (1/100, 11039-1-AP, Proteintech Group, Inc) as described previously [10]. Selected sections were also immunostained with an additional TKT antibody (1/250, H-50, Santa Cruz). Slides were digitally scanned using the NanoZoomer (Hamamatsu Photonics K.K, Hamamatsu, Japan). The intensity of cytoplasmic TKT immunostaining was assessed using a manual scoring method: strong (3+), moderate (2+), weak (1+), or negative (0). A score of 0 or 1+ was defined as low TKT immunostaining and a score or 2+ or 3+ was defined as high TKT immunostaining. The percentage of TKT positive cancer cell nuclei in high power fields (100–200 nuclei) were independently assessed in the ovarian tissues.
Proliferation assays
Proliferation assays were performed using the Xcelligence system (Roche Applied Science, Australia). SKOV-3 cells were grown in 6-well plates at a concentration of 2 × 105 cells/ml overnight and transfected with either TKT silencing siRNA (10 nmol, Ambion), negative siRNA (10 nmol, Ambion), or no siRNA (no treatment, NT). After 48 h, cells were washed, trypsinized and resuspended in antibiotic free RPMI media. SKOV-3 cells at a concentration of 5000 cells/well were added to the E-plates and inserted into the cradle of the Xcelligence system. Continuous impedance was recorded for 96 h to measure cell proliferation in real time. TKT knockdown was confirmed by quantitative RT-PCR and western blotting. All treatments were performed in duplicates and assays were repeated 3–4 times. Additional proliferation experiments were conducted using MTT assays with SKOV-3, OV-90, OVCAR-5, OVCAR-3 and serous primary ovarian cancer cells isolated from patient ascites in the presence of oxythiamine (0–2 mM) (Sigma Aldrich).
Motility and invasion assays
SKOV-3 and OV-90 cells were grown in 6 well-plates at a concentration of 2x105 cells/well overnight and transfected with either TKT silencer siRNA (10 nmol, Ambion, Life Technologies Australia, Mulgrave, Australia) or negative siRNA (10 nmol, Ambion) in 575 μl Opti-MEM media (Invitrogen, Life Tecnhologies Australia) containing 15 μl of Oligofectamine (Invitrogen). An additional control included untreated cells. After 48 h, cells were washed, trypsinized and re-suspended in RPMI media containing 0.1 % BSA. Parallel wells of cells treated with siRNA were re-suspended in RIPA buffer to obtain protein extracts for western blotting to confirm TKT knockdown. Additional cells were treated with increasing concentrations of the TKT inhibitor, oxythiamine (5–1000 µM) for 3 h at room temperature. After treatment with siRNA or oxythiamine, SKOV-3 cells were diluted to a cell concentration of 1 × 106 cells/ml in RPM1 media containing 0.1 % BSA and antibiotics and labelled with calcein-AM (1 μg/ml, Invitrogen) at room temperature for 30 min in dark, on an oscillating platform (Nutator, Clay Adams, Becton–Dickinson, Sparks, MD, USA). Cells were allowed to migrate or invade through uncoated or Geltrex (Invitrogen) coated filters for a further 6 h as described previously [10]. Fluorescence was measured at 485–520 nm using the Triad series multimode detector (Dynex technologies, Chantilly, VA, USA).
Statistical analysis
All analyses were performed using IBM SPSS 21.0 (SPSS Inc., Chicago, IL, USA). For the in vitro cell studies the Student’s t test and one-way ANOVA with Dunnett C or Dunnett T post hoc tests were used to determine statistical significance between control and treatment groups. For the immunohistochemical studies, the Mann–Whitney U, Kruskal–Wallis or Wilcoxon signed rank test were used to determine statistical significance between patient groups. For Kaplan–Meier and Cox regression analyses, relapse or death due to ovarian cancer was used as the endpoint to determine whether TKT levels were associated with progression-free survival or ovarian cancer specific overall survival. Statistical significance was accepted at P < 0.05.
Results
TKT is cleaved by ovarian cancer-peritoneal cell interactions
Protein spots with molecular weights of ~80 and ~43 kDa present in the secretome of co-cultured OVCAR-5 and LP-9 cells but not in the individual cultures were identified to be TKT by mass spectrometry (Fig. 1a, Supplementary Fig. 2). We confirmed by RT-PCR that TKT is expressed by metastatic OVCAR-5, OV-90, and SKOV-3 ovarian cancer cells, by the poorly metastatic OVCAR-3, peritoneal cell line LP-9 as well as primary serous ovarian cancer cells derived from patient ascites (Fig. 1b). Western blotting of cell extracts showed TKT bands at the predicted molecular weight of ~70 kDa in all the cell lines (Fig. 1c).

Transketolase (TKT) is cleaved by ovarian cancer-peritoneal cell interactions, a 2D-gel electrophoresis gel of proteins in the secretome of OVCAR-5 and LP-9 cells. Arrows indicate protein spots identified to be TKT by mass spectrometry that were only present in the ovarian cancer peritoneal co-culture Red proteins present in co-culture, green proteins in mixed single cultures and yellow co-expressed proteins in co-culture and single culture. b TKT expression in ovarian cancer cell lines and primary serous ovarian cancer cells by real-time PCR. Relative TKT expression in ovarian cancer cell lines and primary serous ovarian cancer cells was normalized to the house keeping gene β-actin using the 2−∆∆CT and OVCAR-3 cell expression as a calibrator. c Western blotting confirmed the presence of TKT (70 kDa band in ovarian cancer and LP-9 cell extracts. Recombinant TKT protein was used as the positive control. Equal protein (10 µg) were run on a 10 % SDS-PAGE gel, d CM were collected from single culture or direct co-culture, precipitated with acetone and resuspended in SDS sample buffer. Equal volumes were run on a 10 % SDS-PAGE gel. A smaller TKT band at ~40 kDa was detected in the conditioned media of co-cultured OVCAR-5 and LP-9 cells. Equal volumes were loaded for all samples. TKT processing during OVCAR-5 and LP-9 co-culture was inhibited by treatment with α2 antiplasmin. Western blots in c and d were immunoblotted with rabbit polyclonal TKT antibody (H-50, Santa Cruz Biotechnology)
A single ~70 kDa TKT band was observed in OVCAR-5 cell CM whilst two TKT bands with approximate molecular weights of 70 and 45 kDa were observed in the CM of LP-9 cell alone (Fig. 1d). The molecular weight of rTKT (~120 kDa) was higher than the predicted molecular weight of 68 kDa as it also contained an N-terminal GST-Tag (Fig. 1c). In the CM samples from co-cultured LP-9 and OVCAR-5 cells, three TKT bands were observed; ~70, 50 kDa and a smaller TKT band at ~40 kDa were also present (Fig. 1d). Although smaller in size, the 70 and 40 kDa bands may correspond to the TKT protein spots at 80 and 43 kDa identified in the 2D gel (Fig. 1a). The smaller 40 kDa TKT fragment was also detected when LP-9 cells were co-cultured together with SKOV-3 cells (data not shown). Co-cultured experiments performed in the presence of plasmin inhibitor (α2-antiplasmin) lacked this 40 kDa TKT fragment, confirming that TKT is processed during co-culture (Fig. 1d). The additional band at approximately 50 kDa in the LP-9 and OVCAR-5 co-culture present in Fig. 1d may be an additional cleaved form of TKT as it was not present when cells were treated with anti-plasmin inhibitor. The mass spectrometry data from the ~40 kDa protein spot in the co-cultured CM suggests that N-terminal cleavage of TKT occurs before amino acid 310 (Supplementary Fig. 2).
Nuclear TKT is elevated in peritoneal metastases
Immunohistochemistry showed that ovarian surface epithelium (OSE) and epithelial cells in benign serous cystadenomas exhibited both cytoplasmic and nuclear TKT staining (Fig. 2a, b). Both cytoplasmic and nuclear TKT imunostaining were also observed using an independent TKT antibody (Santa Cruz, H-50, images not shown). Variable cytoplasmic and nuclear TKT positivity was observed in primary serous cancers (Fig. 2c, d). Nuclear TKT positivity in primary serous ovarian cancer tissues (median 82.5 %, range 16.5–100 %) was not significantly different from levels in normal OSE (median 83.3 %, range 34.2–100 %), benign serous tumors (median 90.6 % range 50–100 %) or metastatic tumors (median 88.3 % range 19.7–99.7 %) (Fig. 2f, Kruskal–Wallis, P > 0.05). Cytoplasmic TKT intensity was not altered amongst any of the tissue groups (data not shown). However interestingly, nuclear TKT positivity was increased in peritoneal metastases compared with matching primary cancers (P = 0.01, Wilcoxon Rank test, Fig. 2g). Figure 2d and e show TKT immunostaining in matching primary and metastatic ovarian cancer lesions from the same patient.

TKT expression in human ovarian tissues. TKT immunohistochemistry using rabbit anti-TKT (1:100, Proteintech group) and citrate buffer microwave antigen retrieval. Normal ovary (a), benign serous cystadenoma (b), primary serous cancer (c, d), metastatic lesions (e). d and e are matching tissue samples from the same patients. Bar = 100 µm. f Quantitation of TKT nuclear positivity in ovarian tissues. Normal ovary (n = 6), benign serous tumors (n = 10), primary serous tumors (n = 125), omental metastases (n = 54). g Quantitation of TKT nuclear positivity in matching primary and metastatic peritoneal implants. *, P = 0.01, Wilcoxon Rank test (n = 40)
Nuclear TKT in peritoneal metastases predicts a poorer outcome
Neither cytoplasmic TKT nor nuclear TKT positivity levels analysed as quartiles in the primary cancer were significantly associated with progression-free (PFS) or overall survival (OS) in the Kaplan–Meier analysis (data not shown). When nuclear TKT positivity in peritoneal metastases was separated into quartiles, patients with high nuclear TKT (>94.0 %, 4th quartile, 11/14) had a significantly reduced OS compared to patients with lower TKT positivity (≤94.0 % 1st to 3rd quartiles, 22/40, P = 0.006, Fig. 3a). The 12 month OS rate was 84.2 % in the group of patients with nuclear TKT positivity ≤94 % and only 50.3 % in the group of patients with nuclear TKT positivity >94 %. Nuclear TKT expression in peritoneal metastases was not associated with PFS (Fig. 3b, P = 0.124). Cox regression analysis also indicated that patients with peritoneal metastases with high TKT positivity (>94 %) had a 2.8 fold increased risk of ovarian cancer death (95 % CI; 1.29–5.90, P = 0.009). Cytoplasmic TKT staining in peritoneal metastases was not significantly associated with PFS or OS by Kaplan–Meier analysis (data not shown).

Kaplan Meier survival analysis showing association of nuclear TKT positivity in metastatic lesions with patient outcome. a Advanced serous ovarian cancer patients with high TKT positivity (>94 %, fourth quartile, n = 14) had a significantly reduced overall survival compared to patients with lower TKT positivity (≤94 %, 1st to 3rd quartile, n = 40, P = 0.006, log rank test). b Nuclear TKT positivity in metastatic cancers was not associated with progression-free survival (P = 0.124, log rank test)
TKT knockdown inhibits SKOV-3 cell proliferation
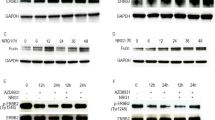
The effect of TKT on the proliferation of metastatic SKOV-3 cells was investigated using two independent TKT siRNA A and B to knockdown TKT expression. Treatment with TKT siRNA A showed a decrease in SKOV-3 cell proliferation over a period of 96 h when compared with negative siRNA treatment or no treatment control (Fig. 4a). Reduced SKOV-3 cell proliferation was also confirmed using TKT siRNA B in an independent experiment (Fig. 4b). TKT siRNA B showed a significant inhibition of proliferation in SKOV-3 cells compared to negative siRNA treatment or NT control at the 24, 48 and 72 h time points (Fig. 4c). The knockdown of TKT in SKOV-3 cells was confirmed by western blotting of cell extracts which showed up to 80 % knockdown of TKT protein following transfection with TKT siRNA A or TKT siRNA B (Fig. 4d).

Effect of TKT siRNA treatment on ovarian cancer cell proliferation, motility and invasion. SKOV-3 proliferation was reduced following transfection with siRNA A (a) and siRNA B (b) over a 96 h time course. c Summary of four independent experiments using siRNA B. siRNA B treatment resulted in a significant reduction in proliferation measured at 24, 48 and 72 h compared to negative siRNA. Data are the mean ± SEM from four independent experiments performed in duplicate. * denotes significant difference from neg siRNA, P < 0.0001, Student t test. d Knockdown of TKT was confirmed by western blotting with a rabbit polyclonal TKT antibody (1:1000, Proteintech). TKT protein was reduced in both SKOV-3 and OV-90 cells following transfection with TKT siRNA A or TKT siRNA B compared to negative siRNA or no treatment controls. TKT siRNA A and B had no effect on the motility or invasion of SKOV-3 (e) or OV-90 cells (f). Data are expressed as percentage of NT control, mean ± SEM from at least 2 independent experiments performed in quadriplicate. P > 0.05, Student t test
Oxythiamine treatment inhibits ovarian cancer cell proliferation
We examined the effects of the TKT inhibitor oxythiamine on ovarian cancer cell proliferation. Increasing concentrations of oxythiamine (0–2 mM) significantly reduced the proliferation of both SKOV-3 and OV-90 cells after 72 h treatment (Fig. 5a). Maximal inhibition was observed using 1 mM oxythiamine for SKOV-3 cells (80 % of control) and 2 mM oxythiamine for OV-90 cells (60 % of control). Oxythiamine (1 mM, 72 h) significantly inhibited the proliferation of SKOV-3, OVCAR-5, OV-90 and OVCAR-3 cells (Fig. 5b). The growth of primary serous ovarian cancer cell lines isolated from ascites (primary 1–8) was also significantly inhibited by oxythiamine (1 mM, 72 h) (Fig. 5c). Importantly, four of these (primary #2, #3, #6 and #7) were derived from patients with chemoresistant disease.

Effect of oxythiamine on ovarian cancer proliferation, motility and invasion. a SKOV-3 and OV-90 cell growth response following 72 h treatment with increasing concentrations of oxythiamine (0–2000 μM) Data are expressed as percentage of control, mean ± SEM from at least three independent experiments performed in triplicate. P > 0.05, Student t test. * denotes significant difference from control. b Oxythiamine (1 mM) significantly inhibits proliferation of SKOV-3, OVCAR-5, OV-90 but not OVCAR-3 after 72 h treatment. c The growth of 8 primary serous ovarian cancer cell lines isolated from ascites (patient # 1–8) was also significantly inhibited by 72 h oxythiamine treatment. Data are expressed as percentage of control, mean ± SEM from at least two independent experiments performed in quadriplicate. P > 0.05, Student t test. * denotes significant difference from control, P < 0.05, Student t test). Oxythiamine treatment had no effect on the motility or invasion of SKOV-3 (d) or OV-90 cells (e). Data are expressed as percentage of control, mean ± SEM from at least two independent experiments performed in quadriplicate P > 0.05, One-Way Anova
TKT knockdown and oxythiamine treatment do not affect ovarian cancer cell motility and invasion
We examined the effects of knocking down TKT expression on ovarian cancer cell motility and invasion in SKOV-3 and OV-90 cells using TKT siRNA A and TKT siRNA B. The knockdown of TKT in both SKOV-3 and OV-90 cells was confirmed by western blotting of cell extracts (Fig. 4d). TKT knockdown did not affect the motility or invasion of SKOV-3 (Fig. 4e) or OV-90 cells (Fig. 4f). Oxythiamine treatment similarly did not affect the motility or invasion of both SKOV-3 and OV-90 cells (Fig. 5d, e).
Discussion
One of the crucial steps in ovarian cancer metastasis involves the implantation of ovarian cancer cells onto the peritoneal lining. To explore the underlying molecular mechanisms, we have recently studied the interaction between ovarian cancer and peritoneal cells in co-culture using a proteomics approach. One of the proteins found to be regulated by ovarian cancer peritoneal interactions is TKT, the key enzyme in the non-oxidative branch of the PPP which mediates the conversion of glucose to ribose phosphate used for nucleotide synthesis. In this study we show that (1) nuclear TKT positivity is increased in peritoneal metastases compared to their matching primary ovarian tumors, (2) high nuclear TKT positivity in peritoneal metastases can predict reduced serous ovarian cancer survival and (3) TKT inhibitors including TKT siRNAs and oxythiamine significantly reduced proliferation of ovarian cancer cell lines and primary serous ovarian cancer cells derived from patient ascites. Together these results encourage further investigation into utilizing TKT inhibitors as novel therapeutics against ovarian cancer.
Most tumors exhibit enhanced glucose metabolism and use anaerobic glycolysis to produce high levels of NADPH and lactate even in the presence of oxygen [21, 22]. This phenomenon first described by Otto Warburg in 1924 [23] has been exploited as a method for tumor detection by using fluorodeoxyglucose positron emission tomography (FDG-PET) [24–26]. Glucose can be degraded via the Embden–Meyerhof glycolytic pathway or an alternate route in the PPP which functions to provide C5 sugars (pentoses) from C6 sugars (hexoses) essential for DNA and RNA synthesis [27]. The thiamine dependent enzyme TKT plays a key role in the non-oxidative branch of PPP pathway and allows oxygen-independent glucose degradation [20, 28–30].
In addition to TKT, the TKT-like proteins, TKTL1 and TKTL2, which share 61 and 66 % amino acid sequence homology with TKT, respectively, are encoded in the human genome [31, 32]. A marked difference between TKT and TKTL1 is a deletion of 38 amino acids in the N-terminal domain (residues 76–113 in TKT) [31, 32]. TKTL1 has been extensively investigated in cancers and has been thought to be a catalytically active mutant form of human TKT [33]. To date, there have been no published studies investigating TKTL2 protein expression in any cancer tissues. Furthermore there is no evidence of its enzymatic activity or reports of its cellular function [34].
TKTL1 is overexpressed in a range of malignancies including breast, colon, gastric, renal, cervical, lung and liver cancers [30, 35–42] and increased TKTL1 levels were shown to be associated with reduced survival for patients with cancers of the colon, oropharynx and the bladder and oral squamous cell carcinomas [30, 38, 43–45]. TKTL1 is elevated in serous ovarian carcinomas compared to other ovarian tumor subtypes [46]. However, an antibody most commonly used to detect TKTL1 (clone JFC12T10) has recently been shown to lack specificity [47, 48]. A re-evaluation using RT-PCR and consecutive sequence analysis identified TKT, not TKTL1, as the dominant isoform of TKTs in a range of cancer cell lines [48]. However it is known that growth conditions can affect TKTL1 expression in cell lines. Hypoxia induces the expression of TKTL1 in human colorectal cancer cells [49] and TKTL1 is activated by hypoxia in glioma cancer cells [50]. As most researcher use normoxic conditions for cultivating cancer cell lines it is likely that the expression of TKTL1 in cancer cell lines may have been underestimated. Although there have been doubts about the specificity of clone JFC12T10, Sun et al. showed a reduction in TKTL1 protein signal intensity by Western blot analysis following TKTL1 knockdown [51]. The specificity of antibody JFC12T10 was also confirmed in cell lines made to stably overexpress TKTL1 [49, 51]. Furthermore independent studies found that TKTL1 mRNA expression correlated with the TKTL1 protein expression [43, 49].
There is also some controversy whether TKTL1 can catalyze the TDP dependant TKT reaction. Computer modeling of TKTL1 revealed marked differences between the structure of TKT and TKT-L1 [52]. The amino acid residues 76–113 missing in TKTL1 form the structure-loop-helix-turn-helix-loop which is necessary for the formation of the active centre of TKT and binding to TDP [52]. A mutant variant of TKT lacking residues 76–113 in the amino terminal region of human TKT could not bind TDP and was devoid of TKT activity [53, 54]. This discovery questioned the role and function of TKTL1 in cancers and its role in glucose metabolism [33]. However both of these studies were conducted in vitro using a mutated TKT protein, not recombinant TKTL1. There is increasingly in vitro and in vivo evidence that TKTL1 exerts a TKT enzyme activity and plays an important role in glucose metabolism and tumor cell proliferation. TKT activity cell was significantly decreased in gastric cancers cells following TKTL1 knockdown [55]. TKTL1 knockdown also significantly reduced the proliferation of gastric and colon cancer cells [55, 56]. Glioma cancer cells were protected from starvation-induced cell death when TKTL1 protein was present [50]. In the absence of functional TKT following TKT knockdown, the stable expression of TKTL1 in leukemia cells was able to replace TKT activity [57].
Compared to TKTL1 there have been only limited studies investigating TKT expression in cancer tissues. A recent study has demonstrated significantly higher TKT expression in pancreatic cancer cell lines compared to TKTL1 and TKTL2 [58]. Although TKT was the predominant expressed gene encoding TKT activity in pancreatic cancer and normal pancreatic ductal cells, higher TKTL1 levels were also seen in pancreatic cancers compared in normal pancreatic ductal cells [58] which is consistent with other cancer studies. TKT expression was also elevated in hepatocellular carcinomas compared to non-tumor tissues [59]. Interestingly, fructose was found to be a potent inducer of TKT expression in pancreatic cancer cells and was preferentially used in the PPP to increase cell proliferation [58]. These findings have not been confirmed in other cancer types but may have major significance for cancer patients, given the high dietary refined fructose consumption in Western diets.
The PPP is up-regulated in cells with anchorage independent cell growth which have high metastatic potential [60]. Tumor cells with an up-regulation of TKT exhibit enhanced non-oxidative glucose usage and therefore are also well adapted to anoxic environments [21] that may be encountered particularly in metastatic sites. In this study, we have demonstrated that nuclear TKT positivity is increased in peritoneal metastases compared to matching primary tumors and high nuclear TKT positivity in peritoneal metastases was associated with reduced ovarian cancer survival. Increased nuclear TKT has also been observed in lung cancer cells isolated from malignant pleural effusions compared to normal bronchiole epithelium [61]. Together these findings suggest that TKT may facilitate survival of metastatic cancer cells by activating the PPP. Glucose metabolic reprogramming of TKT may be important in the pathogenesis of malignant pleural and ascites effusions common in advanced cancers.
The Proteintech rabbit polyclonal antibody used in this study was established by immunizing to the C-terminal 350 amino acid residues of the TKT protein. This region shares 62 % sequence homology to human TKTL1 (accession number P51854.2) and 66 % sequence homology to human TKTL2 (accession number P51854.2). We confirmed specificity to TKT using rTKT and siRNA TKT knockdown. This polyclonal antibody may also detect TKTL1 and TKTL2 as there are several identical amino acid epitopes in common with all three proteins. The other TKT antibody used in this study from Santa Cruz Biotechnology was raised to a 50 amino acid TKT peptide, only has a 44 % amino acid sequence identity with TKTL1 (accession number P51854.2) and 42 % sequence identify with TKTL2 (accession number Q9H19.1). As there is only a 4 amino acid epitope in common with all three proteins, the antibody is unlikely to also detect TKTL1 and TKTL2.
Surprisingly we found TKT to be also present in the nucleus of OSE, benign serous cystadenoma and serous ovarian cancer cells. We verified the nuclear TKT immunostaining using two different TKT antibodies. Both cytoplasmic and nuclear TKT were confirmed to be present in MCF-7 breast cancer cells by immunohistochemistry and western blotting [62]. Medium–high nuclear TKT was also observed in 78 % of normal tissues and 76 % cancer tissues examined in the Human Protein Atlas [63]. As the TKT reactions take place in the cytoplasm, the function of nuclear TKT is not clear at present and warrants further investigation. It is likely that nuclear TKT may have additional functions different from its enzymatic function in the cytoplasm.
TKT was expressed in metastatic (OVCAR-5, SKOV-3, OV-90, primary serous cancer cells present in ascites) and non-metastatic human ovarian cancer cell lines (OVCAR-3) as well as the peritoneal cell line LP-9 by real-time PCR and western blot analysis. TKT was observed at its full length form (70 kDa) in all the cell lines studied, with the highest expression observed by RT-PCR in the highly metastatic ovarian cancer cell line SKOV-3. Interestingly, a smaller fragment of TKT at ~40 kDa was seen in the CM of co-cultured LP-9 and OVCAR-5. This TKT fragment was the same size as observed in the 2D gel with CM from co-cultured LP-9 and SKOV-3 cells (Fig. 1a). Our previous study has identified that plasmin is increased during OVCAR-5 co-culture with LP-9 cells [10]. To confirm that increased plasmin production can lead to TKT processing, we treated co-cultured LP-9 and OVCAR-5 cells in the presence of anti-plasmin inhibitor. We have shown that TKT cleavage as a result of ovarian cancer-peritoneal cell co-culture was blocked by the plasmin inhibitor. TKT fragments have recently been reported in the ascites of ovarian cancer patients [64]. A 35 kDa cleaved TKT fragment has also been identified in the fibroblasts isolated from patients with Alzheimer’s disease [65]. Due to the size of the smaller 40 kDa TKT protein, it is unlikely to exert a TKT enzyme reaction, since invariant, highly conserved amino acid residues are missing. However this smaller TKT isoform could form TKT homodimers or TKT/TKTL1 heterodimers resulting in altered enzymatic kinetics. Furthermore cleavage of TKT-68 kDa full length variant to TKT-40 kDa variant (by plasmin) could lead to variants with higher or lower binding affinity towards TKT or TKTL1. As a consequence normal (two-substrate reaction) TKT activity could be reduced and one-substrate reaction (TKT or TKTL1) could be increased. Further studies are required to investigate the functional role of cleaved TKT in cancer.
Tumors are characterised by enhanced glycolytic rates to meet the high energy requirements for proliferation and growth [66] and increased lipid and nucleotide synthesis to support their biosynthetic needs [14, 67]. Clinical evidence from patients with advanced breast and bronchial carcinoma showed that increased conversion of thiamine to thiamine pyrophosphate in TKT pathways resulted in thiamine deficiency in these patients [68]. Thiamine deficiency frequently occurs in patients with advanced cancer and therefore thiamine supplementation is often used as nutritional support. However, thiamine supplementation may contribute to a high rate of tumor cell survival, proliferation and chemotherapy resistance as doses of thiamine 25 times the recommended daily allowance (RDA) sufficient to correct existing thiamine deficiency were able to stimulate growth of Ehrlich’s ascites tumors [69]. A higher incidence of bladder cancer was also induced in rats fed carcinogen and thiamine compared to rats not supplemented with thiamine [70]. However, very high doses of thiamine (2500-fold RDA) can also inhibit growth of cancer cells [70]. As the effect of thiamine in cancer remains unclear, a cautious approach has been advised before recommending thiamine treatment in patients with malignant disease [71].
TKT has been shown to be a vital factor in supporting proliferation in cells. Vizan et al. [66] showed that TKT enzyme activity increased during late G1 and S phases of cell cycle (during DNA replication) in the human colon cancer cell line HT29, which also coincided with the increased conversion of pentose monophosphates to hexose monophosphates indicating activation of PPP [66]. Therefore, the modification of TKT or its activity could represent a novel approach for developing anti-proliferative cancer treatments. Since this had never been studied in ovarian cancer previously, we used two independent TKT specific siRNAs to knockdown TKT expression in high TKT expressing metastatic SKOV-3 cells. A significant decline in SKOV-3 proliferation was observed with two independent TKT siRNA compared to negative siRNA treatment and no treatment controls. TKT siRNAs have previously also been shown to suppress lung cancer cell proliferation [61].
To further examine the effect of inhibiting TKT enzyme activity on ovarian cell proliferation, SKOV-3, OV-90, OVCAR-5, OVCAR-3 and primary serous ovarian cancer cells isolated from patient ascites were treated with a well-known specific irreversible TKT inhibitor oxythiamine. Oxythiamine treatment at a concentration of 1 mM resulted in a significant decrease in ovarian cancer cell proliferation. This is in agreement with previous studies in several other cancers including malignant pleural effusion cells from patients with lung carcinoma [17, 58, 61, 72]. Boros et al., demonstrated that the inhibition of TKT by oxythiamine resulted in a 90 % decrease in tumor cell mass in Ehrlich ascitic tumors in vivo and inhibition of proliferation of pancreatic adenocarcinoma cells and Ehrlich’s ascitic tumor cells by 39 % [17]. Furthermore, Rais et al., demonstrated that inhibiting TKT reactions by oxythiamine administration resulted in the arrest of Ehrlich’s ascitic tumor cells in the G1 phase of cell cycle [20]. This could be explained by the fact that TKT levels increase during G1/S phase [66]. High daily doses of 500 mg/kg oxythiamine significantly inhibited the proliferation of Lewis Lung carcinoma lung metastases [73]. Oxythiamine treatment could also rescue imatinib resistance in leukemic cells which exhibited increased levels of TKT family genes and increased TKT activity [57].
Neither TKT silencing nor oxythiamine treatment inhibited the motility or invasion of SKOV-3 or OV-90 cells in vitro. In contrast, other researchers studying Lewis Lung Carcinoma cells and using lower concentrations of oxythiamine (20 µM) did not observe an effect on proliferation but a strong inhibition of their invasive ability in vitro by inhibiting the expression of MMP-2 and MMP-9 [73]. Oxythiamine has also been shown to inhibit phosphorylation of 12 proteins involved in cell cycle regulation and apoptosis, particularly Hsp27 which is associated with drug resistance and cell survival in MIA pancreatic cancer cells [72]. TKT can also control secretion of VEGF and vascular permeability in vitro and in vivo [61]. The TKT activator benfotiamine stimulates the activity of PPP enzymes and the phosphorylation/activation of VEGFR2 in a diabetic animal model [74]. Oxythiamine has more recently been shown to alter the dynamics of cellular protein expression in MIA PaCa-2 pancreatic cells by interrupting the rates of de novo protein synthesis involved in several apoptotic signalling pathways [75]. These observations suggest that oxythiamine may affect other signalling pathways in cancer cells. Little is currently known about the safety of oxythiamine and further pre-clinical studies are warranted.
In conclusion, the present study demonstrates that TKT is regulated by peritoneal-ovarian cancer cell interactions and plays an important role in the proliferation of metastatic ovarian cancer cells. These results warrant further studies using TKT inhibitors as potential new chemotherapeutic agents. Inhibiting the TKT pathway that is essential for glucose and fructose metabolism and nucleotide production could be an effective new target for ovarian cancer.
Abbreviations
- CM:
-
Conditioned media
- FBS:
-
Fetal bovine serum
- PPP:
-
Phosphate pentose pathway
- TKT:
-
Transketolase
- TDP:
-
Thiamine diphosphate
References
Jemal A et al (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90
Auersperg N et al (2001) Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev 22(2):255–288
Gardner MJ et al (1995) Expression of cell adhesion molecules on ovarian tumour cell lines and mesothelial cells, in relation to ovarian cancer metastasis. Cancer Lett 91(2):229–234
Freedman RS et al (2004) Peritoneal inflammation—a microenvironment for epithelial ovarian cancer (EOC). J Transl Med 2(1):23
Strobel T, Cannistra SA (1999) Beta1-integrins partly mediate binding of ovarian cancer cells to peritoneal mesothelium in vitro. Gynecol Oncol 73(3):362–367
Ricciardelli C, Rodgers RJ (2006) Extracellular matrix of ovarian tumors. Semin Reprod Med 24(4):270–282
Said NA et al (2007) SPARC inhibits LPA-mediated mesothelial-ovarian cancer cell crosstalk. Neoplasia 9(1):23–35
Heyman L et al (2008) Vitronectin and its receptors partly mediate adhesion of ovarian cancer cells to peritoneal mesothelium in vitro. Tumour Biol 29(4):231–244
Kenny HA et al (2008) The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Investig 118(4):1367–1379
Ween MP et al (2011) Transforming growth factor-beta-induced protein secreted by peritoneal cells increases the metastatic potential of ovarian cancer cells. Int J Cancer 128(7):1570–1584
Lokman NA et al (2013) Annexin A2 is regulated by ovarian cancer-peritoneal cell interactions and promotes metastasis. Oncotarget 4(8):1199–1211
Lindqvist Y et al (1992) Three-dimensional structure of transketolase, a thiamine diphosphate dependent enzyme, at 2.5 A resolution. EMBO J 11(7):2373–2379
Zhao J, Zhong CJ (2009) A review on research progress of transketolase. Neurosci Bull 25(2):94–99
Cascante M et al (2000) Role of thiamin (vitamin B-1) and transketolase in tumor cell proliferation. Nutr Cancer 36(2):150–154
Blass JP, Gibson GE (1977) Abnormality of a thiamine-requiring enzyme in patients with Wernicke–Korsakoff syndrome. N Engl J Med 297(25):1367–1370
Gibson GE et al (1988) Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol 45(8):836–840
Boros LG et al (1997) Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res 57(19):4242–4248
Boros LG et al (2000) Transforming growth factor beta2 promotes glucose carbon incorporation into nucleic acid ribose through the nonoxidative pentose cycle in lung epithelial carcinoma cells. Cancer Res 60(5):1183–1185
Boros LG et al (1998) Inhibition of the oxidative and nonoxidative pentose phosphate pathways by somatostatin: a possible mechanism of antitumor action. Med Hypotheses 50(6):501–506
Rais B et al (1999) Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich’s tumor cells through inhibition of the pentose cycle. FEBS Lett 456(1):113–118
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4(11):891–899
Seyfried TN, Shelton LM (2010) Cancer as a metabolic disease. Nutr Metab 7:7
Warburg O (1956) On the origin of cancer cells. Science 123(3191):309–314
Kelloff GJ et al (2005) Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res 11(8):2785–2808
Kelloff GJ et al (2007) FDG-PET lymphoma demonstration project invitational workshop. Acad Radiol 14(3):330–339
Lavayssiere R, Cabee AE, Filmont JE (2009) Positron emission tomography (PET) and breast cancer in clinical practice. Eur J Radiol 69(1):50–58
Wittig R, Coy JF (2008) The role of glucose metabolism and glucose-associated signalling in cancer. Perspect Med Chem 1:64–82
Boren J et al (2002) Metabolic control analysis aimed at the ribose synthesis pathways of tumor cells: a new strategy for antitumor drug development. Mol Biol Rep 29(1–2):7–12
Ramos-Montoya A et al (2006) Pentose phosphate cycle oxidative and nonoxidative balance: a new vulnerable target for overcoming drug resistance in cancer. Int J Cancer 119(12):2733–2741
Langbein S et al (2006) Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: warburg effect reinterpreted. Br J Cancer 94(4):578–585
Coy JF et al (1996) Molecular cloning of tissue-specific transcripts of a transketolase-related gene: implications for the evolution of new vertebrate genes. Genomics 32(3):309–316
Mitschke L et al (2010) The crystal structure of human transketolase and new insights into its mode of action. J Biol Chem 285(41):31559–31570
Coy JF et al (2005) Mutations in the transketolase-like gene TKTL1: clinical implications for neurodegenerative diseases, diabetes and cancer. Clin Lab 51(5–6):257–273
Kochetov GA, Solovjeva ON (2014) Structure and functioning mechanism of transketolase. Biochim Biophys Acta 9(1844):1608–1618
Staiger WI et al (2006) Expression of the mutated transketolase TKTL1, a molecular marker in gastric cancer. Oncol Rep 16(4):657–661
Foldi M et al (2007) Transketolase protein TKTL1 overexpression: a potential biomarker and therapeutic target in breast cancer. Oncol Rep 17(4):841–845
Zhang S et al (2007) Gene silencing of TKTL1 by RNAi inhibits cell proliferation in human hepatoma cells. Cancer Lett 253(1):108–114
Volker HU et al (2007) Overexpression of transketolase TKTL1 is associated with shorter survival in laryngeal squamous cell carcinomas. Eur Arch Oto-Rhino-Laryngol 264(12):1431–1436
Volker HU et al (2008) Expression of transketolase-like 1 and activation of Akt in grade IV glioblastomas compared with grades II and III astrocytic gliomas. Am J Clin Pathol 130(1):50–57
Langbein S et al (2008) Metastasis is promoted by a bioenergetic switch: new targets for progressive renal cell cancer. Int J Cancer 122(11):2422–2428
Zhang S et al (2008) Overexpression of transketolase protein TKTL1 is associated with occurrence and progression in nasopharyngeal carcinoma: a potential therapeutic target in nasopharyngeal carcinoma. Cancer Biol Ther 7(4):517–522
Chen H et al (2009) Overexpression of transketolase-like gene 1 is associated with cell proliferation in uterine cervix cancer. J Exp Clin Cancer Res 28:43
Kayser G et al (2011) Poor outcome in primary non-small cell lung cancers is predicted by transketolase TKTL1 expression. Pathology 43(7):719–724
Grimm M et al (2013) A biomarker based detection and characterization of carcinomas exploiting two fundamental biophysical mechanisms in mammalian cells. BMC Cancer 13:569
Grimm M et al (2014) GLUT-1(+)/TKTL1(+) coexpression predicts poor outcome in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol 117(6):743–753
Krockenberger M et al (2007) Transketolase-like 1 expression correlates with subtypes of ovarian cancer and the presence of distant metastases. Int J Gynecol Cancer 17(1):101–106
Mayer A, Von Wallbrunn A, Vaupel P (2010) Glucose metabolism of malignant cells is not regulated by transketolase-like (TKTL)-1. Int J Oncol 37(2):265–271
Mayer A, Von Wallbrunn A, Vaupel P (2011) Evidence against a major role for TKTL-1 in hypoxic and normoxic cancer cells. Adv Exp Med Biol 701:123–128
Bentz S et al (2013) Hypoxia induces the expression of transketolase-like 1 in human colorectal cancer. Digestion 88(3):182–192
Wanka C, Steinbach JP, Rieger J (2012) Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J Biol Chem 287(40):33436–33446
Sun W et al (2010) TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res 16(3):857–866
Maslova AO, Meshalkina LE, Kochetov GA (2012) Computer modeling of transketolase-like protein, TKTL1, a marker of certain tumor tissues. Biochem Biokhimiia 77(3):296–299
Meshalkina LE et al (2013) Is transketolase-like protein, TKTL1, transketolase? Biochim Biophys Acta 1832(3):387–390
Schneider S et al (2012) A delta38 deletion variant of human transketolase as a model of transketolase-like protein 1 exhibits no enzymatic activity. PLoS ONE 7(10):e48321
Yuan W et al (2010) Silencing of TKTL1 by siRNA inhibits proliferation of human gastric cancer cells in vitro and in vivo. Cancer Biol Ther 9(9):710–716
Xu X et al (2009) Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int J Cancer 124(6):1330–1337
Zhao F et al (2010) Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene 29(20):2962–2972
Liu H et al (2010) Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res 70(15):6368–6376
Shimizu T et al (2014) Frequent alteration of the protein synthesis of enzymes for glucose metabolism in hepatocellular carcinomas. J Gastroenterol 49(9):1324–1332
Mori S et al (2009) Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 28(31):2796–2805
Lin CC et al (2011) Malignant pleural effusion cells show aberrant glucose metabolism gene expression. Eur Respir J 37(6):1453–1465
Mohd Omar MF et al (2010) Molecular-assisted immunohistochemical optimization. Acta Histochem 112(6):519–528
Uhlen M et al (2010) Towards a knowledge-based human protein atlas. Nat Biotechnol 28(12):1248–1250
Bery A et al (2014) Deciphering the ovarian cancer ascites fluid peptidome. Clin Proteomics 11(1):13
Paoletti F, Mocali A, Tombaccini D (1997) Cysteine proteinases are responsible for characteristic transketolase alterations in Alzheimer fibroblasts. J Cell Physiol 172(1):63–68
Vizan P et al (2009) Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29. Int J Cancer 124(12):2789–2796
Pyragius CE et al (2013) Aberrant lipid metabolism: an emerging diagnostic and therapeutic target in ovarian cancer. Int J Mol Sci 14(4):7742–7756
Basu TK, Dickerson JW (1976) The thiamin status of early cancer patients with particular reference to those with breast and bronchial carcinomas. Oncology 33(5–6):250–252
Comin-Anduix B et al (2001) The effect of thiamine supplementation on tumour proliferation. A metabolic control analysis study. Eur J Biochem FEBS 268(15):4177–4182
Pamukcu AM et al (1970) Effects of the coadministration of thiamine on the incidence of urinary bladder carcinomas in rats fed bracken fern. Cancer Res 30(11):2671–2674
Lu’o’ng KV, Nguyen LT (2013) The role of thiamine in cancer: possible genetic and cellular signaling mechanisms. Cancer Genomics Proteomics 10(4):169–185
Zhang H et al (2010) Inhibition of protein phosphorylation in MIA pancreatic cancer cells: confluence of metabolic and signaling pathways. J Proteome Res 9(2):980–989
Yang CM et al (2010) The in vitro and in vivo anti-metastatic efficacy of oxythiamine and the possible mechanisms of action. Clin Exp Metastasis 27(5):341–349
Ji H et al (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature 448(7155):807–810
Wang J et al (2013) Inhibition of transketolase by oxythiamine altered dynamics of protein signals in pancreatic cancer cells. Exp Hematol Oncol 2:18
Acknowledgments
We thank Dr. Thomas Hamilton (Fox Chase Cancer Center, Philadelphia, PA) for kindly providing the OVCAR-5 cell line, Mrs Wendy Bonner and Dr Noor Hammodi for their help with the TKT immunostaining. This research has been funded by the Ovarian Cancer Research Foundation (OCRF), Australia, Cancer Council of South Australia and South Australian Health and Medical Research Institute.
Conflict of interest
The authors declare they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10585_2015_9718_MOESM1_ESM.tif
Supplementary material 1 (TIFF 3891 kb). Supplementary Fig. 1: Cytokeratin immunocytochemical staining in primary serous ovarian cancer cells. Pan-cytokeratin immunostaining of primary ovarian cancer cells derived from patient 1 (a) and patient 2 (b) ascites compared with the established serous ovarian cancer cell line OV-90 (c). d Represents OV-90 cells negative control of immunocytochemistry without primary antibody. Magnification bar = 100 µm for all images
10585_2015_9718_MOESM2_ESM.tif
Supplementary material 2 (TIFF 798 kb). Supplementary Fig. 2: Ion-trap mass spectrometry LC–MS/MS analysis of spot 1 (a) and spot 2 (b) identified in the secretome of co-cultured OVCAR-5 and LP-9 cells by 2D gel electrophoresis and silver staining
10585_2015_9718_MOESM3_ESM.docx
Supplementary material 3 (DOCX 16 kb). Supplementary Table 1: Summary of clinical and pathological characteristics of the primary ovarian cancer cells established from patient ascites
Rights and permissions
About this article

Cite this article
Ricciardelli, C., Lokman, N.A., Cheruvu, S. et al. Transketolase is upregulated in metastatic peritoneal implants and promotes ovarian cancer cell proliferation. Clin Exp Metastasis 32, 441–455 (2015). https://doi.org/10.1007/s10585-015-9718-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-015-9718-1