
Abstract
Intrachromosomal rearrangements involve a single chromosome and can be formed by several proposed mechanisms. We reported two patients with intrachromosomal duplications and deletions, whose rearrangements and breakpoints were characterized through karyotyping, chromosomal microarray, fluorescence in situ hybridization, whole-genome sequencing, and Sanger sequencing. Inverted duplications associated with terminal deletions, known as inv-dup-del rearrangements, were found in 13q and 15q in these patients. The presence of microhomology at the junction points led to the proposal of the Fold-back mechanism for their formation. The use of different high-resolution techniques allowed for a better characterization of the rearrangements, with Sanger sequencing of the junction points being essential to infer the mechanisms of formation as it revealed microhomologies that were missed by the previous techniques. A karyotype-phenotype correlation was also performed for the characterized rearrangements.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Structural rearrangements are abnormalities in the chromosome structure, which, through an abnormal dosage, rupture, or fusion of genes, can affect the phenotype and cause genomic disorders (Lupski 1998; Gu et al. 2008; Weckselblatt and Rudd 2015).
Among them, intrachromosomal rearrangements involve a single chromosome and may be due to unequal sister chromatid exchanges or recombination between homologous chromosomes (Shaffer and Lupski 2000). Deletions, duplications, inversions, ring chromosomes, inverted duplications associated with terminal deletions (inv-dup-del), and other complex rearrangements are examples of intrachromosomal rearrangements.
Several mechanisms that include DNA recombination, repair, and replication processes (Carvalho and Lupski 2016; Burssed et al. 2022) have been described to explain the formation of structural rearrangements. Non-allelic homologous recombination (NAHR) occurs when there is an unequal crossing-over event after a misalignment that takes place due to the similarity in the sequence of low copy repeats (LCRs) or repetitive elements, such as Alu or long interspersed element-1 (LINE-1), in different parts of the genome (Stankiewicz and Lupski 2002; Shaw and Lupski 2004; Gu et al. 2008; Burssed et al. 2022), resulting in deletions, duplications, and inversions. Non-homologous end-joining (NHEJ) is the main pathway to repair double-strand breaks (DSBs) by bridging, modifying, and then rejoining the broken ends (Gu et al. 2008; Lieber 2008; Burssed et al. 2022).
The formation of inv-dup-del (inverted duplication associated with terminal deletion) rearrangements involves the generation of a dicentric chromosome after (1) a U-type exchange between sister chromatids, or (2) NAHR between inverted LCRs, or (3) recombination inside an inversion loop due to the presence of a paracentric inversion in one of the parents, or (4) a fold-back mechanism due to microhomology (Rowe et al. 2009; Zuffardi et al. 2009; Hermetz et al. 2014; Milosevic et al. 2014; Pedurupillay et al. 2014; Burssed et al. 2022). The last three mechanisms lead to the formation of a disomic spacer, which is a normal copy number region between the two copies of the duplication (Rowe et al. 2009; Burssed et al. 2022). The resulting inv-dup-del chromosome left without telomeres can be stabilized by (1) the addition of a new telomere, creating a simple inverted duplication followed by a terminal deletion, or (2) telomere capture from another chromosome, creating a translocation, or (3) circularization, forming a ring chromosome (Yu and Graf 2010; Burssed et al. 2022).
It is important to study mechanisms of the formation of chromosomal rearrangements as they can assist in understanding cells’ response to damages in their genetic material and the nature of DNA sequences involved in these rearrangements. Signatures of these mechanisms can be found by sequencing the junction points of structural rearrangements, thus allowing us to infer the mechanism for their formation (Weckselblatt and Rudd 2015; Burssed et al. 2022).
Structural rearrangements can be studied through many techniques such as karyotype, chromosomal microarray, fluorescence in situ hybridization (FISH), whole-genome sequencing (WGS), and optical genome mapping (Weckselblatt and Rudd 2015; Mantere et al. 2021; Burssed et al. 2022). These techniques can assist in the characterization of the rearrangement and location of its breakpoints. Sanger sequencing is an essential technique that can confirm breakpoints at the nucleotide level, thus it is used for a more comprehensive analysis and to help in the discovery of the mechanisms (Schluth-Bolard et al. 2013; Moysés-Oliveira et al. 2019; Burssed et al. 2022).
Here, we characterized complex rearrangements in two patients with intrachromosomal simultaneous duplications and deletions, sequenced their breakpoints, inferred the mechanism of formation, and performed a karyotype-phenotype correlation.
Materials and methods
Enrollment
We studied two patients with rearrangements involving intrachromosomal duplications and deletions. The patients were selected and evaluated by the geneticists of the Medical Genetics Center of the Universidade Federal de São Paulo and the São José do Rio Preto Medical School, in Brazil. Patient 2’s parents were also available for the study whereas patient 1’s parents were unavailable. The samples used in this study were collected after written informed consent and approval of the local ethics committee (CAAE 40.846.114.2.0000.5505, CEP 0028/2015).
Karyotype and chromosomal microarray analysis
Peripheral blood was collected into two tubes: one with heparin for the cytogenetic analysis and another one with EDTA for DNA extraction, which was performed using Gentra Puregene Blood Kit (Qiagen-Sciences, MD, USA) according to the manufacturer’s protocol. The DNA quality and quantification were assessed with NanoDrop ND-1000 (Thermo Technologies, Haute-Savoie, France).
G-banding karyotype was performed from lymphocyte cultures according to a modification of the technique from Moorhead et al. (1960). Twenty metaphases were analyzed for each individual and captured with the Ikaros imaging system (Metasystem, Altlussheim, Germany).
Chromosomal microarrays were performed according to the manufacturer’s protocol using different platforms: Genome-Wide Human SNP Array 6.0 for patient 1 and CytoScan 750 K Array for patient 2 and both her parents (Affymetrix, CA, USA). The analysis was carried out using the software ChAS (Affymetrix) using GRCh37/hg19 annotation.
To narrow down the location of the breakpoint, custom chromosomal microarray analysis was performed in both patients using an 8 × 60 K customized slide (Agilent Technologies, CA, USA). Slides were designed with probes distributed in an interval of 10 kb upstream and downstream of the breakpoints detected with the Affymetrix microarray analysis as previously described (Guilherme et al. 2015). Breakpoint analysis was performed using the software Cytogenomics (Agilent).
FISH analysis
Commercial FISH analysis with telomeric probes was performed using Telomere PNA FISH Kit/FITC (Dako Agilent, CA, USA) according to the manufacturer’s protocol. A minimum of five metaphases were analyzed for each individual and the images were captured with the Isis fluorescent imaging system (Metasystem).
Whole-genome sequencing (WGS) analysis
Short-read whole-genome sequencing was performed to help characterize the rearrangement and reach a higher resolution of the breakpoints’ location. The sequencing library was prepared using Nextera™ DNA Flex Library Preparation kit (Illumina, CA, USA) and Nextera™ DNA CD Indexes (Illumina) to generate 600 bp DNA fragments. According to the manufacturer’s protocol, TruSeq PE Cluster Kit v3-cBot-HS and TruSeq Dual Index Sequencing Primer Box (Illumina) were used for clustering, and TruSeq SBS Kit v3-HS (Illumina) was used for sequencing. Paired-end sequencing with 100-bp reads was performed on the HiSeq 2500 platform (Illumina) with an average coverage of 10 × . The BaseSpace platform (Illumina) applications FastQC and BWA Aligner from BaseSpace Labs were used for quality control and alignment, respectively, using default settings. Reads and chimeric inserts were visualized using the software Integrative Genomics Viewer (IGV) 2.4.14 (Broad Institute and the Regents of the University of California) focusing on the regions involved in the rearrangements (Moysés-Oliveira et al. 2019).
Long-range PCR and Sanger sequencing
According to the results from microarray, FISH, and WGS analysis, a region of 1.5 kb surrounding the breakpoints was selected, and primers were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/), and their quality was assessed with the OligoAnalyzer (https://www.idtdna.com/calc/analyzer) tool. Designed primers can be found in Supplementary Table 1 (Hermetz et al. 2014; Guilherme et al. 2015). Long-range PCR was performed using TaKaRa LA Taq® DNA Polymerase kit (Clontech, CA, USA). PCR products were loaded in agarose gel 1%, and the amplification products presenting the expected size were purified with QIAquick® PCR Purification kit (Qiagen) or with QIAquick® Gel Extraction kit (Qiagen). The purified products were submitted to Sanger sequencing to further characterize the junction points. Big dye v3.1 kit (Applied Biosystems, CA, USA) was used for sequencing in Genetic Analyzer 3130xl (Applied Biosystems) and SeqStudio Genetic Analyzer (Applied Biosystems). The electropherograms were analyzed using the software Chromas (Technelysium Pty Ltd, South Brisbane, Australia). The obtained sequences were aligned to GRC37/hg19 using the BLAT tool on the UCSC Genome browser (https://genome.ucsc.edu/).
Karyotype-phenotype correlation
After proper characterization of the rearrangements, annotation and ranking of structural variants, and affected genes were assessed using the AnnotSV tool (Version 3.0), which compiles regulatory and clinically relevant information with data from the Database of Genomic Variants (DGV), Deciphering Developmental Disorders (DDD) Study, Database of Genomic Structural Variation (dbVar), Genome Aggregation Database (gnomAD), Exome Aggregation Consortium (ExAC), Clinical Genome Resource (ClinGen), and Online Mendelian Inheritance in Man (OMIM) databases (Geoffroy et al. 2018). The DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources (DECIPHER, https://decipher.sanger.ac.uk/) as well as previous studies from the literature were used to perform a karyotype-phenotype correlation.
Results
G-banding karyotype and chromosomal microarray revealed the genomic imbalances of the patients with intrachromosomal duplications and deletions (Table 1).
Patient 1
Patient 1 presented neuropsychomotor developmental delay, short stature, facial dysmorphisms (prominent forehead, small eyes, epicanthic folds, long philtrum, thin upper lip), decreased body weight, hypotonia, microcephaly, short and thick neck, micrognathia, multiple palmar creases, overlapping fingers, partial bilateral syndactyly and clinodactyly, central nervous system anomalies (midline malformations, thin corpus callosum, and delay in brain myelination), and several cardiac defects (including single atrium, abnormal atrioventricular and ventriculo-arterial junctions, single valve, mitral and tricuspid atresia, pulmonary valve stenosis, and double outlet left ventricle).
Patient 1’s karyotype (Fig. 1a) result was 46,XX,der(13)dup(13)(q22.3q32.3)del(13)(q32.3q34). Array showed a 22.2 Mb interstitial duplication followed by a 14.0 Mb terminal deletion in the 13q region (Supplementary Fig. 1a). Array (catalog and custom arrays) analysis also showed a normal copy number region between the duplication and the deletion, while FISH using telomeric probes showed that the derivative chromosome presents normal telomeres (Fig. 1b). Whole-genome sequencing assisted in discovering that this patient presents an inv-dup-del rearrangement with the presence of a spacer (Fig. 1c). A chimeric insert in WGS showed that the second copy of the duplication was located in the derivative chromosome 13 in an inverted orientation and that there was a spacer between the two copies of the duplication (Fig. 1d and Table 1). Sanger sequencing revealed the exact location of the breakpoints (chr13:78,927,076 for the start of the duplication, chr13:101,126,244 for the end of the duplication, and chr13:101,128,628 for the start of the deletion) and a 2,384-bp spacer. Two breakpoint junctions can be found in simple inv-dup-del rearrangements: the disomy-inversion, which is between the end of the spacer and the start of the inverted duplication, and the inversion-telomere, which is between the end of the inverted duplication and the neotelomere. At the disomy-inversion junction point, sequencing revealed three nucleotides of microhomology between the end of the spacer and the beginning of the inverted duplication (Fig. 1e). At the inversion-telomere junction point, one nucleotide was added between the end of the inverted duplication and the neotelomere (Fig. 1f).

Results of patient 1. a Partial karyotype at 400-band level resolution showing the normal and the derivative chromosome 13 (arrow). b Telomeric FISH: Metaphase showing the normal chromosome 13 (gray arrows) and the derivative 13 (red arrows) visualized by fluorescence (left) and by inverted banding (right) with telomere signal shown in green. c Ideogram representing the patient’s inv-dup-del rearrangement. To the left, the normal chromosome 13 and the location of the duplication (blue) and the deletion (red). To the right, the altered chromosome 13 with the arrows indicating the orientation of the segments showing the duplicated region (dark blue), the inverted duplication (light blue), and the presence of a spacer (green) between them. d Chimeric insert found in whole-genome sequencing. Inversion in the same chromosome is indicated by aqua-colored reads. Below, a schema highlighting the actual position of the reads in the insert. e Disomy-inversion junction point sequencing. At the top, the electropherogram displaying the spacer (underlined in green) and the inverted duplication (underlined in blue). At the bottom, the alignment of the altered chromosome 13 with the reference sequence of the spacer (green) and the inverted duplication (blue). In purple, the three nucleotides of microhomology. f Inversion-telomere junction point sequencing. At the top, the electropherogram displaying the inverted duplication (underlined in blue), the telomere (underlined in yellow), and the nucleotide addition (orange). At the bottom, the alignment of the altered chromosome 13 with the reference sequence of the inverted duplication (blue) and the telomere (yellow). In orange, the addition of one nucleotide between the two regions. g Interstitial deletion junction point sequencing. At the top, the electropherogram displaying the two ligated regions of the inverted duplication (underlined in different shades of blue) and the region of added nucleotides (underlined in orange). At the bottom, the alignment of the altered chromosome 13 with the reference sequence of the two ligated regions of the inverted duplication (different shades of blue). In orange, the addition of 25 nucleotides between the two regions
The AnnotSV tool revealed 144 genes that were overlapped by the deletion and 110 genes overlapped by the duplication (by at least 1 bp). Based on AnnotSV annotations and published studies, candidate genes to the patient’s phenotypes (ARHGEF7, COL4A1, COL4A2, EFNB2, and ZIC2) were identified. Both alterations were classified as pathogenic (class 5) according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Riggs et al. 2020).
Besides the inv-dup-del rearrangement, the patient also presents a small interstitial deletion of approximately 315 kb located in the duplicated region. Sanger sequencing mapped this deletion at chr13:90,504,388–90,818,809 and showed the addition of 25 nucleotides between the breakpoints (Fig. 1g). The deletion only encompasses one long non-coding RNA and was classified as variant of uncertain significance (VUS) (class 3) according to the ACMG guidelines (Riggs et al. 2020).
Patient 2
Patient 2 presented facial dysmorphisms, underdeveloped nasal alae, micrognathia, hypothyroidism, hearing loss, hydrocephalus, brachydactyly, and mild urinary tract anomalies.
Patient 2’s karyotype (Fig. 2a) result was 46,XX,der(15)dup(15)(q24.3q26.2)del(15)(q26.2q26.3). Array showed a 19.6 Mb interstitial duplication followed by a 5.5 Mb terminal deletion in the 15q region (Supplementary Fig. 1b). Similar to patient 1, custom array analysis also showed a normal copy number region between the duplication and the deletion. FISH using telomeric probes showed that the derivative chromosome presented normal telomeres (Fig. 2b). Observing the karyotype, it was possible to infer that the duplicated copy was located in the derivative chromosome 15. Sanger sequencing allowed us to pinpoint the exact location of the breakpoints (chr15:77,338,547 for the start of the duplication, chr15:96,963,469 for the end of the duplication, and chr15:96,966,212 for the start of the deletion) and revealed that the duplication was in the inverted orientation, thus showing that the patient presents an inv-dup-del rearrangement with the presence of a 2,743-bp spacer (Fig. 2c and Table 1). At the disomy-inversion junction point, sequencing revealed four nucleotides of microhomology between the end of the spacer and the beginning of the inverted duplication (Fig. 2d). At the inversion-telomere junction point, no added nucleotides or microhomologies were found between the end of the inverted duplication and the new telomere (Fig. 2e). From chromosome microarray data, based on single nucleotide polymorphisms that were informative of parent-of-origin within the deleted segment in the patient, we were able to define that the abnormal chromosome originated from the paternal chromosome 15.

Results of patient 2. a Partial karyotype at 400-band level resolution showing the normal and the derivative chromosome 15 (arrow). b Telomeric FISH: Metaphase showing the normal chromosome 15 (gray arrows) and the derivative 15 (red arrows) visualized by fluorescence (left) and by inverted banding (right) with telomere signal shown in green. c Ideogram representing the patient’s inv-dup-del rearrangement. To the left, the normal chromosome 15 and the location of the duplication (blue) and the deletion (red). To the right, the altered chromosome 15 with the arrows indicating the orientation of the segments showing the duplicated region (dark blue), the inverted duplication (light blue), and the presence of a spacer (green) between them. d Disomy-inversion junction point sequencing. At the top, the electropherogram displaying the spacer (underlined in green) and the inverted duplication (underlined in blue). At the bottom, the alignment of the altered chromosome 15 with the reference sequence of the spacer (green) and the inverted duplication (blue). In purple, the four nucleotides of microhomology. e Inversion-telomere junction point sequencing. At the top, the electropherogram displaying the inverted duplication (underlined in blue) and the telomere (underlined in yellow). At the bottom, the alignment of the altered chromosome 15 with the reference sequence of the inverted duplication (blue) and the telomere (yellow)
The AnnotSV tool revealed 44 genes that were overlapped by the deletion and 216 genes overlapped by the duplication (by at least 1 bp). Candidate genes to the patient’s phenotypes (CHSY1, IGF1R) were identified based on AnnotSV annotations and published studies. Both alterations were classified as pathogenic (class 5) according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Riggs et al. 2020).
Discussion
The rearrangements from both patients were characterized by karyotype, Affymetrix catalog array, Agilent custom array, FISH, WGS, and Sanger sequencing, which allowed us to recognize their mechanism of formation.
Mechanisms of formation
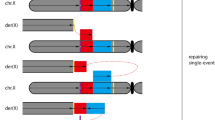
Patient 1 presented an inv-dup-del rearrangement with the presence of a spacer in 13q. From the four most commonly described mechanisms of inv-dup-del formation, the U-type exchange was not responsible since the patient presents the disomic spacer, which is not seen in rearrangements formed by this mechanism (Hermetz et al. 2014; Burssed et al. 2022). The mechanism that relies on NAHR between LCRs was also discarded since, according to UCSC Genome Browser, no LCRs or repetitive elements were present at the breakpoint location. The patient’s parents were not available for evaluation therefore there is no information regarding a possible inversion in their karyotype, which would be needed for the third possible mechanism. The fourth mechanism, named Fold-back, demands the presence of a homologous sequence between the end of the spacer and the beginning of the duplication, and patient 1 exhibits three nucleotides of microhomology in the disomy-inversion junction point. Hermetz et al. (2014) studied inv-dup-dels in 13 patients and verified that the presence of two nucleotides of microhomology is enough for this mechanism to form inv-dup-dels. Although the third mechanism cannot be discarded without evaluation of the parent’s karyotypes, the features identified in the patient’s rearrangement fit very well with the Fold-back mechanism. Therefore, we infer that the Fold-back mechanism was the one responsible for the formation of patient 1’s inv-dup-del rearrangement (Fig. 3). Firstly, a double-strand break caused the terminal deletion of 13q. A subsequent 5′ to 3′ resection created a 3′ single-strand overhang with exposed microhomology that folded back and intrastrand paired with itself in a region of proximal microhomology. DNA replication followed and a dicentric chromosome was formed with the presence of a disomic spacer that corresponds to the loop of the fold back. The dicentric was broken and the inv-dup-del chromosome was formed. Regarding the stabilization of the chromosome end due to the terminal deletion, the inversion-telomere junction point sequencing analysis revealed that the patient presents telomeric sequences (TTAGGG)n which ligate to the end of the inverted duplication through the addition of a single nucleotide (T). These sequences indicate the presence of a neotelomere, which was created by the telomerase enzyme as a healing mechanism (Guilherme et al. 2015). Patient 1 also presents an interstitial smaller deletion located in the duplicated region. Array analysis shows that this region is present in a single copy, which is in the normal chromosome, therefore the deletion is seen within both duplications of the derivative chromosome. If the deletion was located in the normal chromosome, the patient would present two copies of this region, given that the surrounding region is duplicated, and the array result would be normal for this region. Twenty-five nucleotides can be seen ligating both broken ends surrounding the deletion and we can observe that the last 15 nucleotides (ACATTTGCGGTATCT) of the insertion are equal to the following 15 nucleotides in the non-deleted region after the breakpoint, forming a direct repeat, which is a structure commonly seen at the NHEJ junctions (Lieber et al. 2010). Given the nucleotide addition between the breakpoints, and the repeat, we can infer the NHEJ mechanism for the interstitial deletion formation. It is unlikely that this smaller deletion was formed concomitantly with the inv-dup-del rearrangement as it is located far from its breakpoints. The patient’s parents were not available to be studied, however, it is possible that this deletion was inherited.

Fold-back mechanism responsible for the formation of the inv-dup-del rearrangements of patients 1 and 2. a Representation of a chromosome with microhomologies in purple and the centromere in orange. b An initial double-strand break (DSB) generates a terminal deletion. c A 5′ to 3′ resection creates a 3′ overhang with exposed regions of microhomology. d The 3′ overhang can fold back and intrastrand pair with itself at the site of the microhomologies. e DNA synthesis fills the resected gap, creating a monocentric fold-back chromosome. f DNA replication leads to the formation of a dicentric chromosome with a disomic spacer, which corresponds to the fold-back loop region, between the inverted sides of the chromosome. g A second DSB between the two centromeres results in a chromosome with an inverted duplication (blue arrows), a spacer (light green), and a terminal deletion (inv-dup-del)
Patient 2 presented an inv-dup-del rearrangement with the presence of a spacer in 15q. Similar to patient 1, the first two mechanisms of inv-dup-del formation were ruled out due to the presence of a spacer, and the lack of LCRs and repetitive elements at the breakpoint according to UCSC Genome Browser. As for the third mechanism, both parents presented apparently normal karyotypes at 400-band level, not suggesting paracentric inversions. As a result, we infer that the Fold-back mechanism (Fig. 3) is also the one responsible for the formation of this inv-dup-del rearrangement as the disomy-inversion junction point presents four nucleotides of microhomology between the end of the spacer and the beginning of the inverted duplication. The stabilization of the chromosome was also due to the creation of a neotelomere as it was in patient 1 but with the telomeric sequences immediately ligated to the end of the inverted duplication, according to the inversion-telomere junction point sequencing.
It is important to note the importance of breakpoint sequencing in order to infer the correct mechanism of formation of inv-dup-del rearrangements. Hermetz et al. (2014) highlighted that most studies are not able to identify the presence of existing spacers and conclude that the U-type exchange is the most frequent mechanism of inv-dup-del formation. In fact, the Fold-back mechanism has only been described in a few cases in the literature (Hermetz et al. 2014; Milosevic et al. 2014) since many studies use low-resolution techniques when analyzing their patients and, as a result, the microhomologies required for the mechanism are also usually missed.
Karyotype-phenotype correlation
The patient with inv-dup-del(13q) presents two 13q deletions, commonly associated with intellectual disability, growth delay, facial dysmorphisms, hand and foot anomalies, and malformations of the brain, heart, eye, or kidney (Ballarati et al. 2007; Quélin et al. 2009). Patient 1 presents most of these phenotypes therefore it is likely that her 14.0 Mb 13q deletion is responsible for them. Previous studies have associated the haploinsufficiency of 13q genes and regions with certain phenotypes. The ARHGEF7 (Rho guanine nucleotide exchange factor 7, OMIM 605,477) gene has been previously associated with microcephaly (Walczak-Sztulpa et al. 2008). The 13q33.3-13q34 region, which is deleted in the patient, has been associated with congenital heart disease with the collagen IV protein encoded by the COL4A1 (collagen type IV alpha 1 chain, OMIM 120,130) and COL4A2 (collagen type IV alpha 2 chain, OMIM 120,090) genes previously demonstrated to play an essential role in early cardiac development (Schenke-Layland et al. 2011; Hanson et al. 2013). Both these genes were proposed as candidates for heart development in a patient that presented a double outlet right ventricle (Mcmahon et al. 2015), which is a similar phenotype to the patient, who presents a double outlet left ventricle. The reduced expression of the EFNB2 (ephrin B2, OMIM 600,527) gene has also been previously associated with cardiovascular anomalies (Wang et al. 1998; Adams et al. 1999; Cowan et al. 2004). The loss of function of the ZIC2 (zic family member 2, OMIM 603,073) gene has been linked to holoprosencephaly in patients with 13q deletion (Solomon et al. 2010; Quelin et al. 2014). This gene, however, is duplicated in patient 1. Quelin et al. (2014) reported a patient with duplication of ZIC2 that presented cerebral midline malformations, similar to the patient, and suggested that the overexpression of the gene could also be involved with this phenotype. However, this hypothesis has been refuted since Jobanputra et al. (2012) reported a patient with a microduplication encompassing only this gene but without brain malformation. The interstitial 315 kb 13q deletion only encompasses the LINC01049 (Long Intergenic Non-Protein Coding RNA 1049) gene and similarly located deletions were found in three healthy individuals reported in DGV, therefore it does not seem to affect the patient’s phenotype. Other genes and regions in the deleted region or the 22.2 Mb 13q duplication may be responsible for the central nervous system anomalies and other phenotypes that the patient presents.
The patient with inv-dup-del(15q) presents in the 5.5 Mb deleted region the CHSY1 (chondroitin sulfate synthase 1, OMIM 608,183) gene. Li et al. (2010) mapped the Temtamy Preaxial Brachydactyly Syndrome (OMIM #605,282) and identified mutations in the gene in five families with the disease. The syndrome causes symmetric and bilateral preaxial brachydactyly, hyperphalangy, facial dysmorphisms, dental anomalies, neurosensorial hearing loss, and neuro and motor developmental delay. Studies in zebrafish confirmed that the loss of function of this gene, which encodes the chondroitin sulfate synthase 1 protein, essential for chondroitin sulfate biosynthesis, leads to phenotypes equivalent to the ones present in the syndrome, and most of them are present in patient 2. The IGF1R gene (insulin-like growth factor 1 receptor, OMIM 147,370) was also deleted in the patient and DECIPHER patients (#249,298, #261,710, #256,751, #249,789) presenting similar phenotypes such as brachydactyly, clinodactyly, strabismus, telecanthus, microcephaly, wide nasal bridge and small nose, short foot and palm, short stature, hearing impairment, abnormality of the chin, clinodactyly of the 5th finger, underdeveloped nasal alae, and abnormality of the kidney. These deletions involve more genes therefore it is not possible to confirm that IGF1R is the one causing the phenotypes. However, among the genes overlapped by the patient’s deletion, this gene has one of the highest pLI (0, 97) and LOEUF (0, 28) scores, thus indicating that it is extremely intolerant to loss of function. Regarding the patient’s 19.6 Mb duplication, a DECIPHER patient (#253,968) with an overlapping duplication presents hypertelorism, micrognathia, muscular hypotonia, and intellectual deficiency. Another patient (#263,566) also presents a similar duplication and displays macrocephaly, micrognathia, preaxial polydactyly, and delayed speech Given the large size of the duplication, it is likely that the gain in this region’s genes affects the patient’s phenotype. However, no genes annotated by AnnotSV in the duplication were associated with the patient’s phenotypes since they were mainly previously studied as deleted rather than as duplicated.
Conclusion
In this work, two patients had their intrachromosomal rearrangements characterized and their breakpoints mapped using high-resolution techniques. Both patients presented terminal deletions and inverted duplications. The Fold-back mechanism was proposed to form the inv-dup-del rearrangements in 13q and 15q, which were then stabilized with a neotelomere. Sanger sequencing of the junction points was essential to infer the mechanisms of formation as it revealed microhomologies that were missed by the previous techniques.
In patients that present unbalanced non-recurrent rearrangements that involve large regions of the genome, karyotype-phenotype correlation is not simple, seeing as it is difficult to define which gene is causing each phenotype.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- ClinGen :
-
Clinical Genome Resource
- dbVar :
-
Database of Genomic Structural Variation
- DDD :
-
Deciphering Developmental Disorders Study
- DECIPHER :
-
DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources
- DGV :
-
Database of Genomic Variants
- DSBs :
-
Double-strand breaks
- ExAC :
-
Exome Aggregation Consortium
- FISH :
-
Fluorescence in situ hybridization
- gnomAD :
-
Genome Aggregation Database
- LCRs :
-
Low copy repeats
- LINE-1 :
-
Long interspersed element-1
- NAHR :
-
Non-allelic homologous recombination
- NHEJ :
-
Non-homologous end-joining
- OMIM :
-
Online Mendelian Inheritance in Man
- VUS :
-
Variant of uncertain significance
- WGS :
-
Whole-genome sequencing
References
Adams RH et al (1999) Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev 13(3):295–306. https://doi.org/10.1101/gad.13.3.295
Ballarati L et al (2007) 13q Deletion and central nervous system anomalies: further insights from karyotype-phenotype analyses of 14 patients. J Med Genet 44(1):1–6. https://doi.org/10.1136/jmg.2006.043059
Burssed B et al (2022) Mechanisms of structural chromosomal rearrangement formation. Mol Cytogenet 15(1):1–15. https://doi.org/10.1186/s13039-022-00600-6
Carvalho CMB, Lupski JR (2016) Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17(4):224–238. https://doi.org/10.1038/nrg.2015.25
Cowan CA et al (2004) Ephrin-B2 reverse signaling is required for axon pathfinding and cardiac valve formation but not early vascular development. Dev Biol 271(2):263–271. https://doi.org/10.1016/j.ydbio.2004.03.026
Geoffroy V et al (2018) AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34(20):3572–3574. https://doi.org/10.1093/bioinformatics/bty304
Gu W, Zhang F, Lupski JR (2008) Mechanisms for human genomic rearrangements. PathoGenetics 1(1):4. https://doi.org/10.1186/1755-8417-1-4
Guilherme RS et al (2015) Terminal 18q deletions are stabilized by neotelomeres. Mol Cytogenet 8(1):1–7. https://doi.org/10.1186/s13039-015-0135-6
Hanson KP et al (2013) Spatial and temporal analysis of extracellular matrix proteins in the developing murine heart: a blueprint for regeneration. Tissue Engineering - Part A 19(9–10):1132–1143. https://doi.org/10.1089/ten.tea.2012.0316
Hermetz KE et al. (2014) Large inverted duplications in the human genome form via a fold-back mechanism. PLoS Genet 10(1). https://doi.org/10.1371/journal.pgen.1004139
Jobanputra V et al (2012) Duplication of the ZIC2 gene is not associated with holoprosencephaly. Am J Med Genet, Part A 158A(1):103–108. https://doi.org/10.1002/ajmg.a.34375
Li Y et al (2010) Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of BMP signaling. Am J Hum Genet 87(6):757–767. https://doi.org/10.1016/j.ajhg.2010.10.003
Lieber MR (2008) The mechanism of human nonhomologous DNA End joining. J Biol Chem 283(1):1–5. https://doi.org/10.1074/jbc.R700039200
Lieber MR et al (2010) Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell Biochem 50:279–296. https://doi.org/10.1007/978-90-481-3471-7_14
Lupski JR (1998) Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 14(10):417–422. https://doi.org/10.1016/S0168-9525(98)01555-8
Mantere T et al (2021) Optical genome mapping enables constitutional chromosomal aberration detection. Am J Hum Genet 108(8):1409–1422. https://doi.org/10.1016/j.ajhg.2021.05.012
Mcmahon CJ et al (2015) De Novo interstitial deletion 13q33.3q34 in a male patient with double outlet right ventricle, microcephaly, dysmorphic craniofacial findings, and motor and developmental delay. Am J Med Genet A 167(5):1134–1141. https://doi.org/10.1002/ajmg.a.36978
Milosevic J et al (2014) Inverted duplication with deletion: first interstitial case suggesting a novel undescribed mechanism of formation. Am J Med Genet A 164(12):3180–3186. https://doi.org/10.1002/ajmg.a.36777
Moorhead PS et al (1960) Chromosome preparations of leukocytes cultured from human peripheral blood. Exp Cell Res. https://doi.org/10.1016/0014-4827(60)90138-5
Moysés-Oliveira M et al (2019) Breakpoint mapping at nucleotide resolution in X-autosome balanced translocations associated with clinical phenotypes. Eur J Hum Genet 27(5):760–771. https://doi.org/10.1038/s41431-019-0341-5
Pedurupillay CRJ et al (2014) Post-zygotic breakage of a dicentric chromosome results in mosaicism for a telocentric 9p marker chromosome in a boy with developmental delay. Gene 533(1):403–410. https://doi.org/10.1016/j.gene.2013.09.090
Quélin C et al (2009) Twelve new patients with 13q deletion syndrome: genotype-phenotype analyses in progress. Eur J Med Genet 52(1):41–46. https://doi.org/10.1016/j.ejmg.2008.10.002
Quelin C et al (2014) Inversion duplication deletions involving the long arm of chromosome 13: phenotypic description of additional three fetuses and genotype-phenotype correlation. Am J Med Genet A 164(10):2504–2509. https://doi.org/10.1002/ajmg.a.36658
Riggs ER et al (2020) Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22(2):245–257. https://doi.org/10.1038/s41436-019-0686-8
Rowe LR et al (2009) U-type exchange is the most frequent mechanism for inverted duplication with terminal deletion rearrangements. J Med Genet 46(10):694–702. https://doi.org/10.1136/jmg.2008.065052
Schenke-Layland K et al (2011) Recapitulation of the embryonic cardiovascular progenitor cell niche. Biomaterials 32(11):2748–2756. https://doi.org/10.1016/j.biomaterials.2010.12.046
Schluth-Bolard C et al (2013) Breakpoint mapping by next generation sequencing reveals causative gene disruption in patients carrying apparently balanced chromosome rearrangements with intellectual deficiency and/or congenital malformations. J Med Genet. https://doi.org/10.1136/jmedgenet-2012-101351
Shaffer LG, Lupski JR (2000) C Hromosomal R Earrangements in H Umans. Annu Rev Genet 34:297–329. https://doi.org/10.1016/j.ajhg.2009.03.010
Shaw CJ, Lupski JR (2004) Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Human Molecular Genetics 13(REV. ISS. 1):57–64. https://doi.org/10.1093/hmg/ddh073
Solomon BD et al (2010) Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet 154(1):133–141. https://doi.org/10.1002/ajmg.c.30240
Stankiewicz P, Lupski JR (2002) Genome architecture, rearrangements and genomic disorders. Trends Genet 18(2):74–82. https://doi.org/10.1016/S0168-9525(02)02592-1
Walczak-Sztulpa J et al (2008) Chromosome deletions in 13q33–34: report of four patients and review of the literature. Am J Med Genet A 146A(3):337–342. https://doi.org/10.1002/ajmg.a.32127
Wang HU, Chen ZF, Anderson DJ (1998) Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93(5):741–753. https://doi.org/10.1016/S0092-8674(00)81436-1
Weckselblatt B, Rudd MK (2015) Human structural variation: mechanisms of chromosome rearrangements. Trends Genet 31(10):587–599. https://doi.org/10.1016/j.tig.2015.05.010
Yu S, Graf WD (2010) Telomere capture as a frequent mechanism for stabilization of the terminal chromosomal deletion associated with inverted duplication. Cytogenet Genome Res 129(4):265–274. https://doi.org/10.1159/000315887
Zuffardi O et al (2009) Inverted duplications deletions: underdiagnosed rearrangements?? Clin Genet 75(6):505–513. https://doi.org/10.1111/j.1399-0004.2009.01187.x
Acknowledgements
We thank the patients and their families for their participation in the study. This research used facilities of the Brazilian Biorenewables National Laboratory (LNBR), part of the Brazilian Center for Research in Energy and Materials (CNPEM), a private non-profit organization under the supervision of the Brazilian Ministry for Science, Technology, and Innovations (MCTI). The Next Generation Sequencing (NGS) facility staff is acknowledged for their assistance during the experiments of proposal number 25667.
Funding
This work was supported by São Paulo Research Foundation—FAPESP, Brazil (grants #2014/11572–8, #2019/26175–8, and #2019/21644–0) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil.
Author information
Authors and Affiliations
Contributions
BB contributed to the study design, analysis, interpretation of the data, literature review, and wrote the article. MZ contributed to the study design, analysis, and interpretation of the data. BPF contributed to the analysis, interpretation of the data, and critically revised the manuscript. VAM and EMGB contributed to data collection and performed clinical evaluation of patients. FTB contributed to the study design, interpretation of the data, and critically revised the manuscript. MIM contributed to the study design, writing, and critically revised the manuscript. All authors revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval
This study’s protocol was reviewed and approved by the Ethics Committee of the Universidade Federal de São Paulo (CAAE 40846114.2.0000.5505, CEP 0028/2015).
Consent to participate
Written informed consent was obtained from the parents.
Consent for publication
The parents signed informed consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Beth Sullivan
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

10577_2023_9720_MOESM1_ESM.jpg
Supplementary file1 (JPG 3495 kb) Supplementary Fig. 1. Array results. (a) Array results for patient 1 presenting a duplication followed by a terminal deletion in 13p. A small interstitial deletion can be seen within the duplicated region. (b) Array results for patient 2 presenting a duplication followed by a terminal deletion in 15p.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Burssed, B., Zamariolli, M., Favilla, B.P. et al. Fold-back mechanism originating inv-dup-del rearrangements in chromosomes 13 and 15. Chromosome Res 31, 10 (2023). https://doi.org/10.1007/s10577-023-09720-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10577-023-09720-0