
Abstract
Alu elements are a highly successful family of primate-specific retrotransposons that have fundamentally shaped primate evolution, including the evolution of our own species. Alus play critical roles in the formation of neurological networks and the epigenetic regulation of biochemical processes throughout the central nervous system (CNS), and thus are hypothesized to have contributed to the origin of human cognition. Despite the benefits that Alus provide, deleterious Alu activity is associated with a number of neurological and neurodegenerative disorders. In particular, neurological networks are potentially vulnerable to the epigenetic dysregulation of Alu elements operating across the suite of nuclear-encoded mitochondrial genes that are critical for both mitochondrial and CNS function. Here, we highlight the beneficial neurological aspects of Alu elements as well as their potential to cause disease by disrupting key cellular processes across the CNS. We identify at least 37 neurological and neurodegenerative disorders wherein deleterious Alu activity has been implicated as a contributing factor for the manifestation of disease, and for many of these disorders, this activity is operating on genes that are essential for proper mitochondrial function. We conclude that the epigenetic dysregulation of Alu elements can ultimately disrupt mitochondrial homeostasis within the CNS. This mechanism is a plausible source for the incipient neuronal stress that is consistently observed across a spectrum of sporadic neurological and neurodegenerative disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
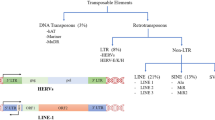
Retrotransposons are mobile genetic elements that utilize an RNA intermediate to copy and paste themselves throughout the genome. There are two primary groups of retrotransposons, those having long-terminal repeats (LTRs) and those without (non-LTR) (Cordaux and Batzer 2009). In the human genome, non-LTR retrotransposons consist of long interspersed elements (LINEs) and short interspersed elements (SINEs), and these collectively account for a remarkable ~ 33% of total genome sequence (Cordaux and Batzer 2009). Alu elements are primate-specific SINEs that are approximately 300 nucleotides in length and are abundant in the human genome, with over 1.3 million elements accounting for at least 11% of overall DNA sequence (Deininger et al. 2003; Hancks and Kazazian 2016). Although once considered to be useless “junk DNA,” the prevalence, diversity, and non-random distribution of Alu elements across primate genomes are suggestive of a functional advantage. Indeed, a large body of evidence documents that Alu elements have directly influenced primate evolution by facilitating genome innovation through novel gene formation, elevated transcriptional diversity, long non-coding RNA and microRNA evolution (including circular RNAs), transcriptional regulation, and creation of novel response elements (Vansant and Reynolds 1995; Norris et al. 1995; Britten 1997; Lev-Maor et al. 2003; Polak and Domany 2006; Laperriere et al. 2007; Lin et al. 2008, 2016; Lehnert et al. 2009; Cordaux and Batzer 2009; Shen et al. 2011; Jeck et al. 2013; Töhönen et al. 2015; Luco 2016; Chen and Yang 2017). Moreover, Alus fundamentally alter the three-dimensional architecture and spatial organization of primate genomes by defining the boundaries of chromatin interaction domains (i.e., topologically associating domains (TADs); Dixon et al. 2012). Genome architecture has a direct influence on biological function, and the observation that Alus are enriched within both TADs and super-enhancer domains (SEDs) supports the hypothesis that Alus directly influence a wide range of critically important processes within primates across multiple levels, from overall genome stability to tissue-specific gene regulation (Huda et al. 2009; Dixon et al. 2012; Soibam 2017; Glinsky 2018). In light of the functional benefits that Alus provide primates, it is interesting to note that Alu retrotransposition events occurred at an estimated 15-fold higher rate in the human, chimpanzee, and bonobo lineage (as compared to other great apes) and a 2.2-fold higher rate in humans when compared to chimpanzee and bonobo (Hedges et al. 2004; Prüfer et al. 2012; Hormozdiari et al. 2013). These evolutionary patterns indicate that positive selection is acting to maintain Alu elements in primate genomes, especially within humans (Mattick and Mehler 2008; Tsirigos and Rigoutsos 2009).
One of the most fascinating and biologically important aspects of Alu elements is that they serve an important role in the formation and function of the brain connectome (Oliver and Greene 2011; Li and Church 2013; Smalheiser 2014; Sakurai et al. 2014; Prendergast et al. 2014; Linker et al. 2017; Bitar and Barry, 2017). Many lines of evidence connect Alu elements with neurogenesis and critical neuronal biochemical processes, including somatic retrotransposition in developing neurons (in parallel to L1 retrotransposition; Baillie et al. 2011; Kurnosov et al. 2015), formation of regulatory circRNAs that are enriched in the central nervous system (CNS) and concentrated at synapses (Jeck et al. 2013; Rybak-Wolf et al. 2015; Chen and Schuman 2016; Floris et al. 2017), regulation of genes that are essential for proper neuron function (e.g., ACE, SMN1, SMN2, SLC6A4; Wu et al. 2013; Ottesen et al. 2017; Schneider et al. 2017), and elevated adenosine-to-inosine (A-to-I) RNA editing in the brain (Mehler and Mattick 2007; Kurnosov et al. 2015; Behm and Öhman 2016). In particular, epigenetic A-to-I editing plays a significant role in mediating neuronal gene expression pathways (Tariq and Jantsch 2012) with Alus serving as the primary target for RNA editing in primates (Picardi et al. 2015; Behm and Öhman 2016). Beyond RNA editing mechanisms, human neuronal gene pathways are regulated by non-coding RNAs originating from Alu elements (e.g., BC200 and NDM29) and specific Alu subfamilies contain retinoic acid response elements which help to regulate neural patterning, differentiation, and axon outgrowth (Vansant and Reynolds 1995; Laperriere et al. 2007; Maden 2007; Castelnuovo et al. 2010; Smalheiser 2014). Moreover, recent discoveries indicate that Alu elements underlie the formation of a vast number of human-specific circRNAs that are hypothesized to play important roles in neurological gene expression pathways (Jeck et al. 2013; Rybak-Wolf et al. 2015; Chen and Schuman 2016; Dong et al. 2017). There is a deep connection between Alus and the formation and function of primate neurological networks, and this has led to the hypothesis that Alu elements were essential for development of the transcriptional diversity and regulation required for the genesis of human cognitive function (Mattick and Mehler 2008; Oliver and Greene 2011; Li and Church 2013; Sakurai et al. 2014).
Despite the functional benefits that Alus have provided primate genomes, Alu elements can disrupt gene expression and function through many pathways (Fig. 1; Deininger and Batzer 1999; Deininger 2011; Tarallo et al. 2012; Ade et al. 2013; Elbarbary et al. 2016; Varizhuk et al. 2016). For this reason, the genome tightly regulates Alus using both DNA methylation and histone (H3K9 methylation) modification in order to control their expression and de novo retrotransposition (Varshney et al. 2015; Elbarbary et al. 2016; Mita and Boeke 2016), and there is mounting evidence indicating that the loss of these epigenetic control mechanisms (due to aging, cellular senescence, environmental factors, and stress) contributes to many forms of cancer, diabetes, osteoporosis, and several mental and neurodegenerative disorders (Szpakowski et al. 2009; Belancio et al. 2010; Muotri et al. 2010; Jintaridth et al. 2013; Dannlowski et al. 2014; Erwin et al. 2014; Bundo et al. 2014; Sun et al. 2014; Goodier 2016; Neven et al. 2016; Bedrosian et al. 2016; Shpyleva et al. 2017; Thongsroy et al. 2017). With respect to deleterious Alu pathways and neurological disease, there are at least 37 mental and neurodegenerative disorders wherein Alu elements are hypothesized to disrupt key cellular processes, thereby resulting in or contributing to the diseased state (Table 1).

Select mechanisms whereby Alu elements can alter gene expression and function (also see Elbarbary et al. 2016). a Sequence homology and orientation of Alu elements contribute to the formation of distinct secondary structures in both DNA and RNA. DNA Alu G-quadruplex structures can alter transcription kinetics (Varizhuk et al. 2016), and pre-mRNA Alu binding forms stem-loop structures that are the primary site for A-to-I editing (see Fig. 2). b Recombination of intra-gene Alu elements resulting in exon deletion. c Exonification of intronic Alus contributing to the production of alternative mRNAs. d Environmental or traumatic stress cascades resulting in increased expression of Alu RNAs that contribute to inflammation (Li and Schmid 2001; Tarallo et al. 2012; Hunter et al. 2015; Lapp et al. 2016), the disruption of global gene transcription through Pol II binding (Mariner et al. 2008), and an increase of H3K9 histone methylation that alters Alu epigenetic pathways (Varshney et al. 2015; Lapp and Hunter 2016; Larsen et al. 2017)
Given the tight connection between Alu elements and the formation and function of the nervous system, it is likely that the dysregulation of Alu elements contributes to many sporadic or idiopathic neurological disorders observed across the global human population (Larsen et al. 2017). Here, we highlight both the beneficial neurological aspects of Alu elements as well as their potential to cause neurological disease. We focus on a novel hypothesis that identifies a potential epigenetic vulnerability to neurological networks that has likely escaped purifying selection. The Alu neurodegeneration hypothesis (sensu Larsen et al. 2017) posits that the epigenetic dysregulation of Alu elements ultimately serves to disrupt mitochondrial homeostasis in neurological networks, thereby setting the stage for increased neuronal stress and neurodegeneration. Given this hypothesis, it is noteworthy that many of the Alu-disrupted genes associated with neurological disorders are related to mitochondrial function and trafficking, including nuclear-encoded mitochondrial genes (i.e., mitonuclear) which help to regulate oxidative stress and metabolic processes in the CNS (Table 1). Mitochondrial dysfunction is implicated across the spectrum of neurological and neurodegenerative disorders that are observed in humans, and this pattern is suggestive of a genetic vulnerability that has evolved in humans. Considering this, we begin by reviewing the integral role that Alu elements have played in human evolution through brain-specific epigenetic A-to-I RNA editing pathways and neurological network formation. Although these Alu-related processes are hypothesized to have contributed to the origin of human cognition, they are likely accompanied by age or stress-related epigenetic vulnerabilities to the CNS, with mitochondrial pathways being especially sensitive.
Alu elements, A-to-I editing, and evolution of the human brain
Alu elements are non-randomly distributed throughout the genome. They occur most frequently within introns and are enriched within genes involved in metabolic, mitochondrial, cellular transport, and binding pathways (Grover et al. 2003; de Andrade et al. 2011; Larsen et al. 2017). Alu nucleotide sequences and lengths (~ 300 bp) are generally conserved (Batzer and Deininger 2002), and it is this seemingly simple aspect of Alu biology that is of monumental biological importance. When inserted within a gene at opposite orientations and at close proximity, Alus bind upon themselves post-transcriptionally, resulting in the formation of a duplex stem-loop structure that is stabilized by the Alu nucleotide sequence and length (Fig. 2; Athanasiadis et al. 2004). These Alu-based secondary structures fundamentally alter the shape of pre-mRNA molecules and serve as the primary binding site for ADAR proteins, which bind to the double-stranded pre-mRNA duplex and edit adenosine (A) residues to inosine (I) thereby recoding pre-mRNAs (Fig. 2). When operating in coding regions (either directly or indirectly), the translation machinery interprets the resulting I residues as guanosine (G) and this mechanism accounts, in part, for the incredible diversity observed in the human proteome that is not encoded within the original DNA sequence (Nishikura 2016). However, the vast majority of A-to-I editing operating on Alu elements occurs within pre-mRNA introns and 3′ UTRs, and this can directly influence gene regulation and function in a surprising number of ways, including the creation of novel splice donor and acceptor sites that result in Alu exonization and alternative gene splicing (Nishikura 2016), recoding of exons immediately adjacent to Alus (Daniel et al. 2014), disruption of RNAi pathways (Chen and Carmichael 2008), production of novel micro-RNA regulatory sites (Borchert et al. 2009), and increased nuclear retention of promiscuously edited mRNAs (Chen and Carmichael 2008 and 2009).

Intronic Alu elements located in close proximity (a) can bind to each other within pre-mRNAs (b) thereby producing a stable stem-loop secondary structure that is the primary substrate for A-to-I editing in primates. ADAR proteins bind to pre-mRNA Alu structures (c) and convert adenosine residues to inosine. If occurring in coding regions, the translation machinery then interprets the inosine residues as guanosine and this can contribute to amino acid changes and alternative protein conformations (d). The model is also applicable to inverted Alus within 3′ untranslated regions, wherein promiscuous editing by ADAR can result in nuclear retention of mRNAs (Chen and Carmichael 2008 and 2009)
Although A-to-I editing plays an essential role in generating transcriptional diversity across eukaryotes, Alu elements provide primate-specific RNA editing opportunities. An example of this is found when comparing the rodent-specific SINE B1 family to Alu. Both B1 and Alu SINE families originated from 7SL RNA (Ullu and Tschudi 1984; Vassetzky et al. 2003), yet rodent-specific B1 elements are approximately half the length (~ 140 bp) of primate Alus (~ 300 bp) and have greater levels of intranucleotide variation. When considering the functional mechanics of A-to-I editing (Fig. 2), the shorter lengths and more variable rodent B1 elements result in the formation of shorter and less-stable double-stranded stem-loop structures in rodent pre-mRNAs. Thus, the molecular properties that separate rodent B1 from primate Alu translate to key functional genomic differences that have influenced evolutionary processes within each lineage (Eisenberg et al. 2005; Neeman et al. 2006; Picardi et al. 2015; Tan et al. 2017).
A-to-I editing associated with Alu elements is perhaps one of the most functionally important yet underappreciated aspect of Alu biology. Approximately 90% of A-to-I editing within primate gene networks centers on Alu elements, and this has fundamentally shaped primate evolution, including the evolution of our own species where A-to-I editing is estimated to occur at over 100 million sites in the human transcriptome (Bazak et al. 2014). Moreover, recent data supports a connection between A-to-I editing and neurological network formation, with elevated editing levels occurring throughout neurogenesis (Behm and Öhman 2016). Genes encoding for key neurological proteins involved in neurotransmission, neurogenesis, gliogenesis, and synaptogenesis are subject to enhanced A-to-I editing, and thus, a number of studies have hypothesized a strong link between A-to-I RNA editing pathways and brain development and function (Schmauss and Howe 2002; Mehler and Mattick 2007; Tan et al. 2009; Sakurai et al. 2014; Liscovitch et al. 2014; Behm and Öhman 2016; Hwang et al. 2016; Picardi et al. 2017b). A recent analysis of A-to-I editing in over 8500 human samples identified tissue-specific editing patterns with elevated editing levels in the brain, including unique patterns in the cerebellum (Tan et al. 2017). The vast majority of these neurologic A-to-I editing events are operating on Alu elements, and, when combined with human-specific Alu evolution (Hedges et al. 2004; Cordaux and Batzer 2009; Prüfer et al. 2012; Hormozdiari et al. 2013), this observation serves as the foundation for the hypothesis that Alu elements and Alu-related pathways contributed to the evolution of enhanced human cognitive abilities (Mattick and Mehler 2008; Li and Church 2013).
Given the relationship between Alu-centric A-to-I editing and the formation and function of the CNS, it is important to expand upon the neuro-specific functions of ADAR proteins. Three ADAR proteins are identified (ADAR1, ADAR2, and ADAR3), and these proteins have distinct tissue-specific expression patterns (Picardi et al. 2015; Tan et al. 2017). ADAR1 and ADAR2 co-opt to regulate neuronal activity by editing key neurotransmitter receptors and ion channels in the CNS (Hood and Emeson 2011). Interestingly, both ADAR2 and ADAR3 have unique brain-specific expression patterns with ADAR2 being highly expressed in the brain and ADAR3 exclusively expressed in the brain (Mehler and Mattick 2007). Until recently, the functional role of the brain-specific ADAR3 protein was largely unknown; however, Oakes et al. (2017) discovered that ADAR3 competes with ADAR2 to regulate glutamate receptor subunit B (GRIA2) A-to-I editing. The GRIA2 protein forms a critical subunit of α-amino-3-hydroxy-5methyl-4-isoxazole propionate (AMPA) receptors, which regulate synaptic calcium and are involved with synaptic plasticity, memory, and learning (Wright and Vissel 2012). Remarkably, A-to-I editing of a specific adenosine nucleotide within GRIA2 results in an amino acid change that alters the GluR-2 protein conformation, thus disrupting calcium permeability of the AMPA receptor and potentially contributing to epilepsy, amyotrophic lateral sclerosis (ALS), and schizophrenia (see Oakes et al. 2017). In light of Oakes et al. (2017), the brain-specific expression pattern of ADAR3 indicates that this protein helps to offset A-to-I editing by ADAR2, perhaps serving to mediate enhanced RNA editing processes throughout the CNS.
Considering the essential role that A-to-I editing processes play in the CNS, the dysregulation of these processes can have a profound impact on the stability of neurological networks (Mehler and Mattick 2007; Rice et al. 2012; Hwang et al. 2016). With respect to ADAR proteins, mutations within ADAR1 have been linked to Aicardi-Goutières syndrome (characterized by severe brain dysfunction; Rice et al. 2012) and Alu-related alternative splicing events of ADAR2 are linked to glioma (Li et al. 2015). Disruption of ADAR1 editing increases production of unedited RNAs which interact with MAV proteins in the outer mitochondrial membrane, ultimately serving to activate inflammatory response pathways (Bajad et al. 2017; Gallo et al. 2017) and perhaps providing a mechanism for inflammatory diseases of the CNS (Hofer and Campbell 2016). ADAR2 knockout mice display epileptic seizures and neuronal death caused by an influx of calcium owing to the disruption of GRIA2 editing (see above). The interference of A-to-I editing processes associated with the KCNA1 gene (encoding a protein essential for potassium regulation and neuron excitability) is hypothesized to underlie episodic ataxia type-1 disorder, a disease of the CNS characterized by seizures, stress-induced ataxia, and myokymia (Ferrick-Kiddie et al. 2017). Moreover, a reduction of A-to-I editing has been observed within hippocampal tissues of Alzheimer’s brains versus healthy controls (Khermesh et al. 2016). From a broader perspective, the disruption of A-to-I editing processes across the CNS has been linked to a wide variety of mental and neurodegenerative disorders including major depression and suicide, epilepsy, schizophrenia, Alzheimer’s disease (AD), and ALS (Gurevich et al. 2002; Kawahara et al. 2004; Kwak and Kawahara 2005; Maas et al. 2006; Kubota-Sakashita et al. 2014; Khermesh et al. 2016; Weissmann et al. 2016; Gal-Mark et al. 2017).
Alu elements, neurogenesis, and the human brain connectome
There is a strong connection between Alu A-to-I editing and the development and function of the brain; therefore, it is impossible to disentangle Alus from the formation and function of neurologic networks (Mehler and Mattick 2007; Tan et al. 2009; Behm and Öhman 2016). It is estimated that the human brain is comprised of over 100 billion neurons that are organized into functional hubs or parcels collectively forming the brain connectome (Van Den Heuvel and Sporns 2013). Beyond major structures of the brain (e.g., cerebellum, frontal cortex, hippocampus, etc.), the existence of connectome parcels shared across unrelated individuals is indicative of an evolutionary conserved process underlying neurological network formation and operating throughout neurogenesis. For example, a recent study mapped the cerebral cortex using multi-modal magnetic resonance imaging and identified 180 connectome parcels that were largely shared across 210 healthy adults (Glasser et al. 2016). Understanding the molecular processes that contribute to the formation of the human brain connectome is essential for understanding the origin of human-specific neurological disorders and diseases observed across the global distribution of our species. This is especially true for neurodegenerative conditions that are hypothesized to originate in functional network hubs and progress along neuronal network connections (e.g., AD; Seeley 2017; Cope et al. 2018).
A growing body of evidence indicates that retrotransposons (including both LINEs and SINEs) are active throughout neurogenesis and contribute to mosaic neuron genomes that ultimately form the human brain connectome (Muotri et al. 2005; Erwin et al. 2014; Kurnosov et al. 2015; Evrony 2016; Paquola et al. 2016; Linker et al. 2017). Although somatic L1 retrotransposition events within developing neurons have received much attention, it is noteworthy that Alu retrotransposition occurs in parallel with L1 throughout neurogenesis (Baillie et al. 2011; Kurnosov et al. 2015), thus providing primate-specific aspects of neurologic network formation. Furthermore, there is evidence that unites Alu elements with retinoic acid regulation (Vansant and Reynolds 1995; Laperriere et al. 2007), which is essential for neuronal patterning and differentiation throughout neurogenesis and is a potential regulator of neuron regeneration (Maden 2007). Retinoic acid is vital for the establishment, maintenance, and repair of neuronal networks, and given the presence of retinoic acid response elements in Alu elements, it is possible that Alu activity during neurogenesis is connected to retinoic acid signaling processes.
Considering the Alu regulatory pathways discussed above, it is of great interest to note that retrotransposition of Alu elements is hypothesized to occur at elevated levels within the dentate gyrus of the hippocampus, the putative site of adult neurogenesis (Kurnosov et al. 2015). Moreover, A-to-I editing levels steadily increase as neural progenitor cells develop into adult neurons (Behm and Öhman 2016). These data indicate that at least two retrotransposon-centric processes (somatic retrotransposition of both LINES and SINES and enhanced A-to-I editing operating primarily on Alu elements) are major contributors to neurogenesis, perhaps serving to establish the neuronal and biochemical diversity that underlies the ~ 100 billion neuron brain connectome. Remarkably, emerging data suggests that a third Alu-centric process is associated with the formation and function of neurological networks, this being the production of circRNAs that are enriched in the brain and concentrated at synaptic junctions (Jeck et al. 2013; Rybak-Wolf et al. 2015; Chen and Schuman 2016). Identifying vulnerabilities to each of these retrotransposon-centric processes will likely contribute to the identification of novel mechanisms underlying mental disorders and neurologic disease and could lead to novel therapeutic interventions.
Pathways to incipient neuronal stress and neurological disease
The disruption of Alu-centric epigenetic RNA editing processes is implicated across the entire spectrum of neurologic disorders (see above). In light of this observation, it is interesting to note that another, seemingly unrelated, feature of many neurological disorders is mitochondrial dysfunction (Lin and Beal 2006; Rugarli and Langer 2012; Gottschalk et al. 2014; Petschner et al. 2017). However, we have previously shown that mitonuclear genes are enriched with Alu elements when compared to random (Larsen et al. 2017), which is consistent with earlier observations regarding the non-random insertion of Alu elements into genes associated with transcriptionally active regions of the genome (Grover et al. 2003; de Andrade et al. 2011). Thus, it is likely that Alu-mediated gene regulatory processes are actively influencing mitonuclear gene expression, regulation, and protein function through the pathways discussed above and reviewed in Chen and Yang (2017). Knowing this, the dysregulation of epigenetic Alu regulatory pathways is a plausible source for mitochondrial stress and dysfunction, with the CNS being particularly vulnerable (Larsen et al. 2017). Such a mechanism could contribute to the initial activation of complex mitochondrial stress pathways and incipient neuronal stress associated with sporadic neurologic disorders (e.g., inflammation, immune response, mitophagy, etc.). Importantly, these processes would precede macroscopic pathologies such as protein aggregation and neuronal atrophy observed in neurodegenerative diseases (Swerdlow et al. 2010; Larsen et al. 2017; Swerdlow 2017).
The Alu neurodegeneration hypothesis (sensu Larsen et al. 2017) proposes a “double-edged sword,” whereby the beneficial Alu-related processes that underlie neuron diversity and function also have the potential to disrupt mitochondrial homeostasis across neurological networks through deleterious cascade events that are facilitated by eroding tissue-specific Alu epigenetic control mechanisms. The stability of the brain’s connectome and the entire CNS depends on healthy mitochondrial populations within neurons, astrocytes, microglia, and supporting cells (Cai et al. 2011; Viader et al. 2011; Schwarz 2013; Jackson and Robinson 2017). Mitochondria play critical roles for a wide range of essential neuronal processes including glucose and lipid metabolism, metal ion biosynthesis, cellular trafficking along axons, neurotransmitter relay across synapses, and synaptic calcium homoeostasis (Schwarz 2013; Harbauer et al. 2014). Therefore, molecular mechanisms that are known to disrupt gene expression and protein folding of genes that are essential for mitochondrial function can ultimately disrupt neurological function.
Interference of mitochondrial dynamics across the CNS is consistently hypothesized to occur during the earliest stages of mental, neurological, and neurodegenerative disorders ranging from depression, epilepsy, and schizophrenia to ALS, AD, and Parkinson’s disease (PD; Lu 2009; Rezin et al. 2009; Kim et al. 2010; Coskun et al. 2012; Martin 2012; Gottschalk et al. 2014; Larsen et al. 2017; Flippo and Strack 2017; Petschner et al. 2017). Collectively, these disorders are estimated to impact approximately 250 million people globally, accounting for at least 10.2% of the global disease burden (GBD 2015 Neurologic Disorders Collaborator Group 2017). The occurrence of sporadic forms of human-specific neurologic disorders (e.g., non-familial schizophrenia, ALS, late-onset AD, PD, etc.) across the entire distribution of our species is suggestive of a common yet complex genetic mechanism that evolved in primates and is amplified in humans (Larsen et al. 2017). Considering this, we expand on the mitocentric view of idiopathic neurologic disease manifestation by reviewing the evidence that unites primate-specific Alu activity with incipient neurologic mitochondrial dysfunction.
Eukaryotic mitochondria are hypothesized to have originated from an endosymbiotic alphaproteobacterium which, over expansive evolutionary time, evolved in parallel with host genomes into the mitochondrial organelles that we observe today (Roger et al. 2017). The human mitochondrial genome encodes only 13 proteins, yet it is estimated that human mitochondria depend on approximately ~ 2000 genes encoded within the nuclear genome for their functionality (Calvo et al. 2015; Johnson et al. 2017). These mitonuclear genes are thus subject to deleterious Alu activity, and Alu-related deleterious events have been linked to many neurologic and neurodegenerative disorders, including epilepsy, Wilson’s disease, Leigh syndrome, PD, ALS, and AD (Table 1 and references therein; Fig. 3). When considering the incipient mitochondrial dysfunction observed across the spectrum of neurological neurodegenerative disorders, it is possible that tissue-specific epigenetic dysregulation of Alu elements within the CNS can ultimately manifest into distinct disease phenotypes (Larsen et al. 2017).

Deleterious Alu activity operating on mitonuclear genes can disrupt mitochondrial function in the CNS and contribute to a number of diseased phenotypes (see Table 1). The type and severity of associated neurological and neurodegenerative disorders depends on the deleterious Alu mechanism of action, the mitonuclear gene pathways involved, the time or developmental stage of induction, level or severity of traumatic stress, and tissue specificity (see Larsen et al. 2017). If operating across the suite of mitonuclear genes through epigenetic pathways, the mechanism helps to explain the origin of incipient mitochondrial stress and CNS connectome destabilization that is observed across the spectrum of neurological and neurodegenerative disorders
Several interesting patterns emerge when examining the key neurologic processes that are disrupted through deleterious Alu activity (Table 1). For example, mitochondria play an essential role in maintaining intracellular metal ion homeostasis (e.g., iron, copper, and zinc), the disruption of which can result in the increased production of free radicals that damage mitochondria and contribute to the increased production of reduced oxygen species (ROS; Rossi et al. 2004; Madsen and Gitlin 2007). The brain is especially sensitive to ROS production, and iron, copper, and zinc-related oxidative stress has been linked to many neurodegenerative disorders including AD, PD, and Wilson’s disease (Rossi et al. 2004; Madsen and Gitlin 2007). It is notable that deleterious Alu activity has been identified in several genes that are essential to maintaining proper iron and copper homeostasis, including FXN, ATP7A, ATP7B, HMBS, NDUFS2, SLC30A6, and PARK7 (DJ-1) (Table 1; Gu et al. 2007; Kaler 2011; Girotto et al. 2014). Knowing this, it is possible that either global or tissue-specific dysregulation of Alu elements within mitonuclear genes can alter mitochondrial metal ion processing pathways thereby contributing to increased ROS production leading to neurologic stress.
A second interesting pattern with respect to deleterious neurologic Alu activity concerns metabolic pathways. The efficient processing of glucose and lipids across the CNS is critical for the stability and function of neurons, and the disruption of mitochondrial-mediated metabolic pathways has been linked to many neurologic disorders including AD and peripheral neuropathies (Viader et al. 2013; De La Monte and Tong 2014). Deleterious Alu activity occurs in genes that are critical for glucose and lipid metabolism, including ABCD1, ACAT1, ALMS1, APOB, GK, GLA, HPRT, LPL, PDHA1, PMM2, PSEN1, SOD2, and SPAST (Table 1). Several of these genes encode for mitochondrial-related proteins that have been implicated in metabolic diseases that directly, or indirectly, contribute to neurological dysfunction. The connection between Alu elements and metabolic pathways is consistent with the observation that Alu elements preferentially insert into metabolic genes, and this has led to the hypothesis that Alus regulate the expression of genes related to type 1 diabetes (Grover et al. 2003; Mirza et al. 2014; Kaur and Pociot 2015). Moreover, Alu RNAs act to suppress the expression of both endothelial nitric oxide synthase (eNOS) and superoxide dismutase 2 (SOD2) during hyperglycemic conditions (Wang et al. 2016), suggesting a regulatory role of Alu elements during oxidative stress and strengthening the link between Alu element activity and diabetes.
There is growing evidence linking sporadic AD with dysfunctional metabolic pathways, leading some to consider AD as a “type 3 diabetes” wherein glycolysis and lipid homeostasis are altered (Steen et al. 2005; De La Monte et al. 2006; De La Monte and Wands 2008; De La Monte and Tong 2014; De Felice and Lourenco 2015; Mittal et al. 2016). The most well-documented risk factor for AD is a variant within APOE (APOE ε4), a gene which encodes for a glycoprotein that mediates cholesterol and lipid transport (Saunders et al. 1993; Strittmatter et al. 1993; Mahley and Rall 2000). The APOE ε4 allele is strongly associated with earlier onset of AD, and it is hypothesized that this is a result of the disruption of cholesterol processing and subsequent accumulation of amyloid precursor proteins (APP; i.e., the amyloid cascade hypothesis). Although the “amyloid cascade hypothesis” has dominated Alzheimer’s research for decades (Hardy and Higgins 1992; Selkoe 2000; McKhann et al. 2011), the failure of multiple drug trials targeting amyloid pathways has led many in the Alzheimer’s research community to search for alternative hypotheses that can help explain the origin of neurodegenerative disease as well as novel molecular pathways with therapeutic potential (Herrup 2015).
It is of particular interest then to note that a second genetic risk factor for AD, TOMM40, is located immediately adjacent to APOE on human chromosome 19, and the two genes are in tight linkage disequilibrium (Lyall et al. 2014; Roses et al. 2016a). TOMM40 encodes for a beta-barrel protein that ultimately forms a central pore in the outer mitochondrial membrane (Shiota et al. 2015), and much like APOE, genetic variants of TOMM40 are linked to cognitive impairment and neurodegenerative disease (Roses 2010; Gottschalk et al. 2014; Greenbaum et al. 2014; Payton et al. 2016; Roses et al. 2016b; Arpawong et al. 2017). The most well known of these TOMM40 variants is the rs10524523 (rs523) homopolymer repeat, a variable stretch of deoxythymidine (T) located within TOMM40 intron 6 (Roses 2010). The rs523 poly-T varies in length from approximately 12 to 46 nucleotides, and the longer variants are statistically associated with thinning of the hippocampus (independent of the APOE ε4 allele; Burggren et al. 2017) and earlier onset of AD (Lutz et al. 2010; Roses et al. 2010). Interestingly, rs523 is embedded within tandemly repeated Alu elements and originated from an Alu insertion event (Payton et al. 2016; Larsen et al. 2017). At least 149 Alu A-to-I editing events are identified within TOMM40, the majority of which are associated with Alu elements surrounding the rs523 repeat and intron 9 (Picardi et al. 2017a).
There is a potentially important link that unties APOE APP processing with the functional mechanics of pre-protein transport through the TOMM pore. It is possible that conformational changes of the Tom40 protein, potentially originating from Alu-mediated events (see above, reviewed in Elbarbary et al. 2016; Chen and Yang 2017; Larsen et al. 2017), can ultimately serve to restrict the passage of lipids across the outer mitochondrial membrane (Larsen et al. 2017). When combined with altered APP processing, this process could account for the initial site of intracellular protein accumulation that is hypothesized to precede extracellular plaque formation during very early stages of AD (Skovronsky et al. 1998; D’Andrea et al. 2001; Takahashi et al. 2002). Consistent with this hypothesis is the direct observation of APP accumulation at the TOMM pore (Devi et al. 2006) as well as functional tolerance of Tom40 conformational changes by mitochondria (Mager et al. 2011; Kuszak et al. 2015). Importantly, this mechanism could help to explain the common patterns of protein accumulation (e.g., amyloid plaques and alpha-synuclein Lewy bodies) observed across the spectrum of neurodegenerative disease, including ALS, AD, and PD (Ross and Poirier 2004; Gottschalk et al. 2014; Larsen et al. 2017). An age or stress-related component to Tom40 conformational changes comes with the epigenetic dysregulation of Alu elements associated with the aging process or traumatic stress (see Larsen et al. 2017). Whether or not these processes are directly mediated by deleterious Alu events remains to be tested; however, it is notable that Alu exons and Alu somatic retrotransposition events have been identified in several TOM genes that are required for the stability of the translocase of the outer mitochondrial membrane and pre-protein import, including TOMM5, TOMM7, TOMM22, TOMM40, and TOMM40L (Baillie et al. 2011; de Andrade et al. 2011; Lin et al. 2016).
With respect to Alu elements, mitochondrial dysfunction, and the broader pathological scope of AD and other neurodegenerative diseases, there is evidence suggesting that Alu-derived peptides interact with tau proteins, perhaps serving a regulatory role for tau phosphorylation (Hoenicka et al. 2002). Tau is a microtubule-associated protein that functions to stabilize axonal microtubules and to transport mitochondria along axons, and taupathies (including tau hyperphosphorylation) are a characteristic feature of several neurodegenerative diseases including AD, progressive supranuclear palsy, corticobasal degeneration, and Pick’s disease (Ittner and Götz 2011; Khanna et al. 2016). The MAPT gene encodes for tau, and alternative splicing events of MAPT result in multiple tau isoforms (Reddy 2011). Approximately 86 Alu elements (including FLAMs) are distributed throughout MAPT introns, and A-to-I editing is occurring at 315 Alu-related sites with elevated levels at the 3′ end of MAPT (REDIportal database; Picardi et al. 2017a). When considering the potential for Alu structural variants of MAPT (including DNA and pre-mRNA secondary structures) and evidence of Alu RNAs interacting with tau proteins (Hoenicka et al. 2002), we recommend additional studies aimed at elucidating the regulatory impacts that Alu elements might have on MAPT gene expression and tau phosphorylation.
A-to-I editing and the potential for mitochondrial stress
Although several neurological disorders are hypothesized to be the result of disruptive A-to-I editing processes across the CNS (see above), it is presently unknown whether or not these processes are actively influencing mitochondrial function. What evidence is there indicating that post-transcriptional modification of mitonuclear genes can alter gene expression or function? Are there particular neurological or neurodegenerative disorders that are associated with mitonuclear genes that have elevated levels of A-to-I editing? To provide insights into these questions, we searched the REDIportal A-to-I editing database (Picardi et al. 2017a) for mitonuclear genes where (1) A-to-I editing has been identified within Alu elements in coding regions and (2) A-to-I editing has contributed to non-synonymous amino acid changes. We identified 57 mitonuclear genes with A-to-I editing occurring within putative Alu exons, and in 52 of these genes, the post-transcriptional modification resulted in non-synonymous amino acid changes (Supplementary Table 1). Many of these genes are involved with essential neuronal processes including calcium binding and transport, zinc transport, apoptosis regulation, voltage-gated ion channels, and mitochondrial elongation with notable examples including ADSL, BAX, CASP2, COQ2, DFFB, FBXO18, LYRM4, PACRG, and SLC30A6 (Supplementary Table 1).
From a broader perspective, we identified enhanced A-to-I editing across 134 mitonuclear genes that are associated with a spectrum of neurologic and neurodegenerative disorders ranging from depression, tobacco use disorder, and bipolar disorder to ALS, Leigh syndrome, PD, and AD (Supplementary Table 2). In light of these patterns, we hypothesize that system-wide or tissue-specific epigenetic dysregulation of Alu A-to-I editing within the CNS can serve to disrupt key mitochondrial biochemical processes, thus potentially contributing to incipient mitochondrial and neuronal stress (Fig. 3).
Conclusions
Enhanced somatic retrotransposon throughout neurogenesis contributes to the mosaic brain; however, such activity likely contributes to mosaic pathways leading to disease (Erwin et al. 2014). Elucidating these pathways might ultimately provide insight into the sporadic nature of idiopathic diseases that are impacting the global human population. The disruption of Alu-mediated pathways that underlie gene regulation is a plausible mechanism for the origin of complex human-specific neurologic and neurodegenerative disorders. Although many of these disorders have similar phenotypes (e.g., mitochondrial dysfunction), it is possible that these phenotypes arise from deleterious activity operating across tissue-specific gene networks. If correlated with eroding or fluctuating epigenetic control mechanisms of retrotransposons that are associated with aging, cellular senescence, and/or cellular stress (Belancio et al. 2010; Pal and Tyler 2016; Schneider et al. 2017), then such mechanisms might largely escape purifying selection and would be difficult to detect using traditional methods (e.g., genome-wide association studies). It is important to note that the Alu-centric mechanisms discussed herein collectively provide a unified framework for multiple hypotheses that have been put forth regarding the origin of neurodegenerative disease including inflammation, oxidative stress, metabolic dysfunction, and accumulation of protein bodies (see above).
Alu elements have played a pivotal role in the evolution of the human epigenome (Prendergast et al. 2014), and both hypermethylation and hypomethylation of Alu elements have been correlated with a number of age-related disorders including Alzheimer’s disease, multiple sclerosis, osteoporosis, and many forms of cancer (Bollati et al. 2009; Jintaridth and Mutirangura 2010; Belancio et al. 2010; Jintaridth et al. 2013; Neven et al. 2016). In light of these patterns, as well as the newly discovered regulatory roles of Alu elements (Polak and Domany 2006; Chen and Carmichael 2008; Chen and Yang 2017), we recommend additional research that focuses on the epigenetic interplay between Alu elements and mitochondrial gene networks in the central nervous system.
Abbreviations
- A-to-I:
-
Adenosine-to-inosine
- AD:
-
Alzheimer’s disease
- ADAR:
-
Adenosine deaminase acting on RNA
- ALS:
-
Amyotrophic lateral sclerosis
- AMPA:
-
α-Amino-3-hydroxy-5methyl-4-isoxazole propionate
- APP:
-
Amyloid precursor protein
- circRNAs:
-
Circular RNAs
- CNS:
-
Central nervous system
- FLAM:
-
Free left Alu monomer
- LINE:
-
Long interspersed element
- L1:
-
Long interspersed element-1
- LTR:
-
Long-terminal repeat
- mRNA:
-
Messenger RNA
- PD:
-
Parkinson’s disease
- pre-mRNA:
-
Precursor messenger RNA
- SEDs:
-
Super-enhancer domains
- SINE:
-
Short interspersed element
- TADs:
-
Topologically associating domains
- TOMM:
-
Translocase of outer mitochondrial membrane
References
Ade C, Roy-Engel AM, Deininger PL (2013) Alu elements: an intrinsic source of human genome instability. Curr. Opin. Virol. 3:639–645
Antonell A, Gelpi E, Sánchez-Valle R et al (2012) Breakpoint sequence analysis of an AβPP locus duplication associated with autosomal dominant Alzheimer’s disease and severe cerebral amyloid angiopathy. J Alzheimers Dis 28:303–3088
Arnoldi A, Tonelli A, Crippa F et al (2008) A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat 29:522–531
Arpawong TE, Pendleton N, Mekli K et al (2017) Genetic variants specific to aging-related verbal memory: insights from GWASs in a population-based cohort. PLoS One 12:e0182448
Athanasiadis A, Rich A, Maas S (2004) Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2:e391
Baillie JK, Barnett MW, Upton KR et al (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479:534–537
Bajad P, Jantsch MF, Keegan L, O’Connell M (2017) A to I editing in disease is not fake news. RNA Biol 14:1223–1231
Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nat Rev Genet 3:370–379
Bazak L, Haviv A, Barak M et al (2014) A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24:365–376
Bedrosian TA, Linker S, Gage FH (2016) Environment-driven somatic mosaicism in brain disorders. Genome Med 8:58
Behm M, Öhman M (2016) RNA editing: a contributor to neuronal dynamics in the mammalian brain. Trends Genet 32:165–175
Belancio VP, Roy-Engel AM, Deininger PL (2010) All y’all need to know ’bout retroelements in cancer. Semin Cancer Biol 20:200–210
Bhattacharjee A, Yang H, Duffy M et al (2016) The activity of menkes disease protein ATP7A is essential for redox balance in mitochondria. J Biol Chem 291:16644–16658
Bitar M, Barry G (2017) Multiple innovations in genetic and epigenetic mechanisms cooperate to underpin human brain evolution. Mol Biol Evol msx303. https://doi.org/10.1093/molbev/msx303
Bollati V, Schwartz J, Wright R et al (2009) Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev 130:234–239
Bondurand N, Fouquet V, Baral V et al (2012) Alu-mediated deletion of SOX10 regulatory elements in Waardenburg syndrome type 4. Eur J Hum Genet 20:990–994. https://doi.org/10.1038/ejhg.2012.29
Bondy-Chorney E, Crawford Parks TE, Ravel-Chapuis A et al (2016) Staufen1 regulates multiple alternative splicing events either positively or negatively in DM1 indicating its role as a disease modifier. PLoS Genet 12:e1005827. https://doi.org/10.1371/journal.pgen.1005827
Bonifati V, Rizzu P, van Baren MJ et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259. https://doi.org/10.1126/science.1077209
Boone PM, Yuan B, Campbell IM et al (2014) The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. Am J Hum Genet 95:143–161
Borchert GM, Gilmore BL, Spengler RM et al (2009) Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet 18:4801–4807. https://doi.org/10.1093/hmg/ddp443
Bouchet C, Vuillaumier-Barrot S, Gonzales M et al (2007) Detection of an Alu insertion in the POMT1 gene from three French Walker Warburg syndrome families. Mol Genet Metab 90:93–96. https://doi.org/10.1016/j.ymgme.2006.09.005
Britten RJ (1997) Mobile elements inserted in the distant past have taken on important functions. Gene 205:177–182
Brooks EM, Branda RF, Nicklas JA, O’Neill JP (2001) Molecular description of three macro-deletions and an Alu-Alu recombination-mediated duplication in the HPRT gene in four patients with Lesch-Nyhan disease. Mutat Res 476:43–54
Bundo M, Toyoshima M, Okada Y et al (2014) Increased L1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81:306–313. https://doi.org/10.1016/j.neuron.2013.10.053
Burggren AC, Mahmood Z, Harrison TM et al (2017) Hippocampal thinning linked to longer TOMM40 poly-T variant lengths in the absence of the APOE ε4 variant. Alzheimer’s Dement 13:739–748. https://doi.org/10.1016/j.jalz.2016.12.009
Cai Q, Davis ML, Sheng Z-H (2011) Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci Res 70:9–15. https://doi.org/10.1016/j.neures.2011.02.005
Calvo SE, Clauser KR, Mootha VK (2015) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44:D1251–D1257. https://doi.org/10.1093/nar/gkv1003
Castelnuovo M, Massone S, Tasso R et al (2010) An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. FASEB J 24:4033–4046. https://doi.org/10.1096/fj.10-157032
Checler F, Goiran T, da Costa AC (2017) Presenilins at the crossroad of a functional interplay between PARK/PARKIN and PINK1 to control mitophagy: implications for neurodegenerative diseases. Autophagy 15:1–2. https://doi.org/10.1080/15548627.2017.1363950
Chen L-L, Carmichael GG (2008) Gene regulation by SINES and inosines: biological consequences of A-to-I editing of Alu element inverted repeats. Cell Cycle 7:3294–3301. https://doi.org/10.4161/cc.7.21.6927
Chen L-L, Carmichael GG (2009) Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Molecular Cell 35:467–478
Chen W, Schuman E (2016) Circular RNAs in brain and other tissues: a functional enigma. Trends Neurosci 39:597–604. https://doi.org/10.1016/j.tins.2016.06.006
Chen L-L, Yang L (2017) ALUternative regulation for gene expression. Trends Cell Biol 27:480–490. https://doi.org/10.1016/j.tcb.2017.01.002
Choi B-O, Kim NK, Park SW et al (2011) Inheritance of Charcot–Marie–Tooth disease 1A with rare nonrecurrent genomic rearrangement. Neurogenetics 12:51–58. https://doi.org/10.1007/s10048-010-0272-3
Chong-Chong X, Denton KR, Wang Z et al (2016) Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis Model Mech 9:39–49
Conceição Pereira M, Loureiro JL, Pinto-Basto J et al (2012) Alu elements mediate large SPG11 gene rearrangements: further spatacsin mutations. Genet Med 14:143–151. https://doi.org/10.1038/gim.2011.7
Cope TE, Rittman T, Borchert RJ, et al. (2018) Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy. Brain, awx347, doi.org:10.1093/brain/awx347
Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10:691–703. https://doi.org/10.1038/nrg2640
Coskun P, Wyrembak J, Schriner SE et al (2012) A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta (BBA)-General Subj 1820:553–564
D’Andrea MR, Nagele RG, Wang HY et al (2001) Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 38:120–134. https://doi.org/10.1046/j.1365-2559.2001.01082.x
Daniel C, Silberberg G, Behm M, Öhman M (2014) Alu elements shape the primate transcriptome by cis-regulation of RNA editing. Genome Biol 15:R28. https://doi.org/10.1186/gb-2014-15-2-r28
Dannlowski U, Kugel H, Redlich R et al (2014) Serotonin transporter gene methylation is associated with hippocampal gray matter volume. Hum Brain Mapp 35:5356–5367. https://doi.org/10.1002/hbm.22555
Dawson G, Kilkus J, Siakotos AN et al (1995) Mitochondrial abnormalities in CLN2 and CLN3 forms batten disease. Mol Chem. Neuro 29:227–235
De Andrade A, Wang M, Bonaldo MF et al (2011) Genetic and epigenetic variations contributed by Alu retrotransposition. BMC Genomics 12:617
De Felice FG, Lourenco MV (2015) Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer’s disease. Front Aging Neurosci 7:94. https://doi.org/10.3389/fnagi.2015.00094
De La Monte SM, Tong M (2014) Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 88:548–559
De La Monte SM, Wands JR (2008) Alzheimer’s disease is type 3 diabetes—evidence reviewed. J Diabetes Sci Technol 2:1101–1113. https://doi.org/10.1177/193229680800200619
De La Monte SM, Tong M, Lester-Coll N et al (2006) Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis 10:89–109. https://doi.org/10.2165/11597760-000000000-00000
Deininger P (2011) Alu elements: know the SINEs. Genome Biol 12:1
Deininger PL, Batzer MA (1999) Alu repeats and human disease. Mol Genet Metab 67:183–193
Deininger PL, Moran JV, Batzer MA, Kazazian HH (2003) Mobile elements and mammalian genome evolution. Curr Opin Genet Dev 13:651–658
Devi L, Prabhu BM, Galati DF et al (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26:9057–9068
Dixon JR, Selvaraj S, Yue F et al (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380
Dobrovolny R, Nazarenko I, Kim J et al (2011) Detection of large gene rearrangements in X-linked genes by dosage analysis: identification of novel α-galactosidase A (GLA) deletions causing Fabry disease. Hum Mutat 32:688–695. https://doi.org/10.1002/humu.21474
Dong R, Ma XK, Chen LL, Yang L (2017) Increased complexity of circRNA expression during species evolution. RNA Biol 14:1064–1074
Eisenberg E, Nemzer S, Kinar Y et al (2005) Is abundant A-to-I RNA editing primate-specific? Trends Genet 21:77–81. https://doi.org/10.1016/j.tig.2004.12.005
Elbarbary RA, Lucas BA, Maquat LE (2016) Retrotransposons as regulators of gene expression. Science 351:aac7247
Erwin JA, Marchetto MC, Gage FH (2014) Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci 15:497–506
Evrony GD (2016) One brain, many genomes. Science 354:557–558
Ferrari L, Scuvera G, Tucci A et al (2017) Identification of an atypical microdeletion generating the RNF135-SUZ12 chimeric gene and causing a position effect in an NF1 patient with overgrowth. Hum Genet 136:1329–1339. https://doi.org/10.1007/s00439-017-1832-5
Ferrick-Kiddie EA, Rosenthal JJC, Ayers GD, Emeson RB (2017) Mutations underlying Episodic Ataxia type-1 antagonize Kv1.1 RNA editing. Sci Rep 7:41095. https://doi.org/10.1038/srep41095
Flippo KH, Strack S (2017) An emerging role for mitochondrial dynamics in schizophrenia. Schizophr. Res. 187:26–32
Floris G, Zhang L, Follesa P, Sun T (2017) Regulatory role of circular RNAs and neurological disorders. Mol Neurobiol 54:156–5165
Gallo A, Vukic D, Michalík D et al (2017) ADAR RNA editing in human disease; more to it than meets the I. Hum Genet 136:1265–1278. https://doi.org/10.1007/s00439-017-1837-0
Gallus GN, Cardaioli E, Rufa A et al (2010) Alu-element insertion in an OPA1 intron sequence associated with autosomal dominant optic atrophy. Mol Vis 10:178–183
Gal-Mark N, Shallev L, Sweetat S et al (2017) Abnormalities in A-to-I RNA editing patterns in CNS injuries correlate with dynamic changes in cell type composition. Sci Rep 7:43421. https://doi.org/10.1038/srep43421
GBD 2015 Neurological Disorders Collaborator Group 2015 NDC (2017) Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol 16:877–897. https://doi.org/10.1016/S1474-4422(17)30299-5
Girotto S, Cendron L, Bisaglia M et al (2014) DJ-1 is a copper chaperone acting on SOD1 activation. J Biol Chem 289:10887–10899. https://doi.org/10.1074/jbc.M113.535112
Glasser MF, Coalson TS, Robinson EC et al (2016) A multi-modal parcellation of human cerebral cortex. Nature 536:171–178. https://doi.org/10.1038/nature18933
Glinsky GV (2018) Contribution of transposable elements and distal enhancers to evolution of human-specific features of interphase chromatin architecture in embryonic stem cells. Chromosome Res https://doi.org/10.1007/s10577-018-9571-6
Goodier JL (2016) Restricting retrotransposons: a review. Mob DNA 7:16. https://doi.org/10.1186/s13100-016-0070-z
Gottschalk WK, Lutz MW, He YT et al (2014) The broad impact of TOM40 on neurodegenerative diseases in aging. J Parkinsons Dis Alzheimers Dis 1
Greenbaum L, Springer RR, Lutz MW et al (2014) The TOMM40 poly-T rs10524523 variant is associated with cognitive performance among non-demented elderly with type 2 diabetes. Eur Neuropsychopharmacol 24:1492–1499
Grover D, Majumder PP, Rao CB, et al (2003) Nonrandom distribution of Alu elements in genes of various functional categories: insight from analysis of human chromosomes 21 and 22. Mol Biol Evol 20:1420–1424. doi: https://doi.org/10.1093/molbev/msg153
Gu Y, Kodama H, Watanabe S et al (2007) The first reported case of Menkes disease caused by an Alu insertion mutation. Brain Dev 29:105–108. https://doi.org/10.1016/j.braindev.2006.05.012
Gu S, Yuan B, Campbell IM et al (2015) Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum Mol Genet 24:4061–4077. https://doi.org/10.1093/hmg/ddv146
Gurevich I, Tamir H, Arango V et al (2002) Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron 34:349–356. https://doi.org/10.1016/S0896-6273(02)00660-8
Hancks DC, Kazazian HH (2016) Roles for retrotransposon insertions in human disease. Mob DNA 7(1)
Harbauer AB, Zahedi RP, Sickmann A et al (2014) The protein import machinery of mitochondria—a regulatory hub in metabolism, stress, and disease. Cell Metab 19:357–372
Hardy J, Higgins G (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science (80- ) 256:184–185. https://doi.org/10.1126/science.1566067
Hedges DJ, Callinan PA, Cordaux R et al (2004) Differential Alu mobilization and polymorphism among the human and chimpanzee lineages. Genome Res 14:1068–1075
Herrup K (2015) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18:794–799. https://doi.org/10.1038/nn.4017
Hoenicka J, Arrasate M, de Yebenes JG, Avila J (2002) A two-hybrid screening of human Tau protein: interactions with Alu-derived domain. Neuroreport 13:343–349
Hofer MJ, Campbell IL (2016) Immunoinflammatory diseases of the central nervous system—the tale of two cytokines. Br J Pharmacol 173:716–728. https://doi.org/10.1111/bph.13175
Hood JL, Emeson RB (2011) Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Springer, Berlin, Heidelberg, pp 61–90
Hormozdiari F, Konkel MK, Prado-Martinez J et al (2013) Rates and patterns of great ape retrotransposition. Proc Natl Acad Sci USA 110:13457–13462. https://doi.org/10.1073/pnas.1310914110
Huang LS, Ripps ME, Korman SH et al (1989) Hypobetalipoproteinemia due to an apolipoprotein B gene exon 21 deletion derived by Alu-Alu recombination. J Biol Chem 264:11394–11400
Huda A, Mariño-Ramírez L, Landsman D, Jordan IK (2009) Repetitive DNA elements, nucleosome binding and human gene expression. Gene 436:12–22
Hunter R, Gagnidze K, McEwen B, Pfaff D (2015) Stress and the dynamic genome: steroids, epigenetics, and the transposome. Proc Natl Acad Sci USA 112:6828–6833
Hwang T, Park C-K, Leung AKL et al (2016) Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19:1093–1099. https://doi.org/10.1038/nn.4337
Ittner LM, Götz J (2011) Amyloid-β and tau—a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci 12:65–72. https://doi.org/10.1038/nrn2967
Jackson JG, Robinson MB (2017) Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia. https://doi.org/10.1002/glia.23252
Jeck WR, Sorrentino JA, Wang K et al (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19:141–157. https://doi.org/10.1261/rna.035667.112
Jiang J, Wu X, Shen D et al (2017) Analysis of RP2 and RPGR mutations in five X-linked Chinese families with retinitis pigmentosa. Sci Rep 7:44465. https://doi.org/10.1038/srep44465
Jintaridth P, Mutirangura A (2010) Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics 41:194–200
Jintaridth P, Tungtrongchitr R, Preutthipan S, Mutirangura A (2013) Hypomethylation of Alu elements in post-menopausal women with osteoporosis. PLoS One 8:e70386. https://doi.org/10.1371/journal.pone.0070386
Jobic F, Morin G, Vincent-Delorme C et al (2017) New intragenic rearrangements in non-Finnish mulibrey nanism. Am J Med Genet Part A 173:2782–2788. https://doi.org/10.1002/ajmg.a.38381
Johnson SC, Gonzalez B, Zhang Q et al (2017) Network analysis of mitonuclear GWAS reveals functional networks and tissue expression profiles of disease-associated genes. Hum Genet 136:55–65. https://doi.org/10.1007/s00439-016-1736-9
Kaler SG (2011) ATP7A-related copper transport diseases—emerging concepts and future trends. Nat Rev Neurol 7:15–29. https://doi.org/10.1038/nrneurol.2010.180
Kaneko H, Dridi S, Tarallo V et al (2011) DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471:325–330. https://doi.org/10.1038/nature09830
Kaur S, Pociot F (2015) Alu elements as novel regulators of gene expression in type 1 diabetes susceptibility genes? Genes (Basel) 6:577–591. https://doi.org/10.3390/genes6030577
Kawahara Y, Ito K, Sun H et al (2004) Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801. https://doi.org/10.1038/427801a
Khanna MR, Kovalevich J, Lee VMY et al (2016) Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimer’s Dement. 12:1051–1065
Khermesh K, D’Erchia AM, Barak M et al (2016) Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer’s disease. RNA 22:290–302. https://doi.org/10.1261/rna.054627.115
Kim J, Moody JP, Edgerly CK et al (2010) Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet 19:3919–3935
Kim Y, Tarallo V, Kerur N et al (2014) DICER1/Alu RNA dysmetabolism induces caspase-8-mediated cell death in age-related macular degeneration. Proc Natl Acad Sci U S A 111:16082–16087. https://doi.org/10.1073/pnas.1403814111
Knight Johnson A, Schaefer GB, Lee J et al (2017) Alu-mediated deletion of PIGL in a Patient with CHIME syndrome. Am J Med Genet Part A 173:1378–1382. https://doi.org/10.1002/ajmg.a.38181
Kubota-Sakashita M, Iwamoto K, Bundo M, Kato T (2014) A role of ADAR2 and RNA editing of glutamate receptors in mood disorders and schizophrenia. Mol Brain 7:5. https://doi.org/10.1186/1756-6606-7-5
Kurnosov AA, Ustyugova SV, Nazarov VI et al (2015) The evidence for increased L1 activity in the site of human adult brain neurogenesis. PLoS One 10:e0117854. https://doi.org/10.1371/journal.pone.0117854
Kuszak AJ, Jacobs D, Gurnev PA et al (2015) Evidence of distinct channel conformations and substrate binding affinities for the nitochondrial outer membrane protein translocase pore Tom40. J Biol Chem 290:26204–26217. https://doi.org/10.1074/jbc.M115.642173
Kutsche K, Ressler B, Katzera H et al (2002) Characterization of breakpoint sequences of five rearrangements in L1CAM and ABCD1 (ALD) genes. Hum Mutat 19:526–535
Kwak S, Kawahara Y (2005) Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J Mol Med (Berl) 83:110–120. https://doi.org/10.1007/s00109-004-0599-z
Laperriere D, Wang T-T, White JH, Mader S (2007) Widespread Alu repeat-driven expansion of consensus DR2 retinoic acid response elements during primate evolution. BMC Genomics 8:23. https://doi.org/10.1186/1471-2164-8-23
Lapp HE, Hunter RG (2016) The dynamic genome: transposons and environmental adaptation in the nervous system. Epigenomics 8:237–249
Larsen PA, Lutz MW, Hunnicutt KE et al (2017) The Alu neurodegeneration hypothesis: a primate-specific mechanism for neuronal transcription noise, mitochondrial dysfunction, and manifestation of neurodegenerative disease. Alzheimer’s Dement. 13:828–838
Le Guennec K, Veugelen S, Quenez O et al (2017) Deletion of exons 9 and 10 of the presenilin 1 gene in a patient with early-onset Alzheimer disease generates longer amyloid seeds. Neurobiol Dis 104:97–103. https://doi.org/10.1016/j.nbd.2017.04.020
Lehnert S, Van Loo P, Thilakarathne PJ et al (2009) Evidence for co-evolution between human microRNAs and Alu-repeats. https://doi.org/10.1371/journal.pone.0004456
Lerner T, Boustany RMN, Anderson JW et al (1995) Isolation of a novel gene underlying Batten disease, CLN3. Cell 82:949–957
Lev-Maor G, Sorek R, Shomron N, Ast G (2003) The birth of an alternatively spliced exon: 3’splice-site selection in Alu exons. Science 300:1288–1291
Li JB, Church GM (2013) Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat Neurosci 16:1518–1522. https://doi.org/10.1038/nn.3539
Li TH, Schmid CW (2001) Differential stress induction of individual Alu loci: implications for transcription and retrotransposition. Gene 276:135–141
Li W, Jin Y, Prazak L et al (2012) Transposable elements in TDP-43-mediated neurodegenerative disorders. PLOS One:e44099, 1–e44010
Li Z, Tian Y, Tian N et al (2015) Aberrant alternative splicing pattern of ADAR2 downregulates adenosine-to-inosine editing in glioma. Oncol Rep 33:2845–2852. https://doi.org/10.3892/or.2015.3907
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Lin L, Shen S, Tye A et al (2008) Diverse splicing patterns of exonized Alu elements in human tissues. PLoS Genet 4:e1000225. https://doi.org/10.1371/journal.pgen.1000225
Lin L, Jiang P, Park JW et al (2016) The contribution of Alu exons to the human proteome. Genome Biol 17:15
Linker SB, Marchetto MC, Narvaiza I et al (2017) Examining non-LTR retrotransposons in the context of the evolving primate brain. BMC Biol 15:68. https://doi.org/10.1186/s12915-017-0409-z
Liscovitch N, Bazak L, Levanon EY, Chechik G (2014) Positive correlation between ADAR expression and its targets suggests a complex regulation mediated by RNA editing in the human brain. RNA Biol 11:1447–1456. https://doi.org/10.4161/15476286.2014.992286
López E, Casasnovas C, Giménez J et al (2015) Characterization of Alu and recombination-associated motifs mediating a large homozygous SPG7 gene rearrangement causing hereditary spastic paraplegia. Neurogenetics 16:97–105. https://doi.org/10.1007/s10048-014-0429-6
Lu B (2009) Mitochondrial dynamics and neurodegeneration. Curr Neurol Neurosci Rep 9:212
Luco RF (2016) Retrotransposons jump into alternative-splicing regulation via a long noncoding RNA. Nat Struct Mol Biol 23:952–954. https://doi.org/10.1038/nsmb.3318
Lutz MW, Crenshaw DG, Saunders AM, Roses AD (2010) Genetic variation at a single locus and age of onset for Alzheimer’s disease. Alzheimer’s Dement 6:125–131
Lyall DM, Harris SE, Bastin ME et al (2014) Alzheimer’s disease susceptibility genes APOE and TOMM40, and brain white matter integrity in the Lothian Birth Cohort 1936. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2014.01.006
Maas S, Kawahara Y, Tamburro KM, Nishikura K (2006) A-to-I RNA editing and human disease. RNA Biol 3:1–9
Maden M (2007) Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 8:755–765. https://doi.org/10.1038/nrn2212
Madsen E, Gitlin JD (2007) Copper and iron disorders of the brain. Annu Rev Neurosci 30:317–337. https://doi.org/10.1146/annurev.neuro.30.051606.094232
Mager F, Gessmann D, Nussberger S, Zeth K (2011) Functional refolding and characterization of two Tom40 isoforms from human mitochondria. J Membr Biol 242:11–21
Mahley RW, Rall SC (2000) Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 1:507–537. https://doi.org/10.1146/annurev.genom.1.1.507
Mameli E, Lepori MB, Chiappe F et al (2015) Wilson’s disease caused by alternative splicing and Alu exonization due to a homozygous 3039-bp deletion spanning from intron 1 to exon 2 of the ATP7B gene. 569:276–279
Mariner PD, Walters RD, Espinoza CA et al (2008) Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol Cell 29:499–509
Martin LJ (2012) Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci 107:355–415. https://doi.org/10.1016/B978-0-12-385883-2.00005-9
Mattick JS, Mehler MF (2008) RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci 31:227–233. https://doi.org/10.1016/j.tins.2008.02.003
McKhann GM, Knopman DS, Chertkow H et al (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 7:263–269
Mefford HC, Zemel M, Geraghty E et al (2015) Intragenic deletions of ALDH7A1 in pyridoxine-dependent epilepsy caused by Alu-Alu recombination. Neurology 85:756–762. https://doi.org/10.1212/WNL.0000000000001883
Mehler MF, Mattick JS (2007) Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev 87:799–823
Mirza AH, Kaur S, Brorsson CA, Pociot F (2014) Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS One. https://doi.org/10.1371/journal.pone.0105723
Mita P, Boeke JD (2016) How retrotransposons shape genome regulation. Curr Opin Genet Dev 37:90–100. https://doi.org/10.1016/j.gde.2016.01.001
Mittal K, Mani RJ, Katare DP (2016) Type 3 diabetes: cross talk between differentially regulated proteins of type 2 diabetes mellitus and Alzheimer’s disease. Sci Rep 6:25589. https://doi.org/10.1038/srep25589
Morais S, Bastos-Ferreira R, Sequeiros J, Alonso I (2016) Genomic mechanisms underlying PARK2 large deletions identified in a cohort of patients with PD. Neurol Genet 2:e73. https://doi.org/10.1212/NXG.0000000000000073
Muotri AR, Chu VT, Marchetto MCN et al (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435:903–910
Muotri AR, Marchetto MCN, Coufal NG et al (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446. https://doi.org/10.1038/nature09544
Mustajoki S, Ahola H, Mustajoki P, Kauppinen R (1999) Insertion of Alu element responsible for acute intermittent porphyria. Hum Mutat 13:431–438
Neeman Y, Levanon EY, Jantsch MF, Eisenberg E (2006) RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 12:1802–1809. https://doi.org/10.1261/rna.165106
Neven KY, Piola M, Angelici L et al (2016) Repetitive element hypermethylation in multiple sclerosis patients. BMC Genet 17:84. https://doi.org/10.1186/s12863-016-0395-0
Nishikura K (2016) A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17:83–96. https://doi.org/10.1038/nrm.2015.4
Norris J, Fan D, Aleman C et al (1995) Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J Biol Chem 270:22777–22782. https://doi.org/10.1074/JBC.270.39.22777
Nozu K, Iijima K, Ohtsuka Y et al (2014) Alport syndrome caused by a COL4A5 deletion and exonization of an adjacent AluY. Mol Genet genomic Med 2:451–453
Oakes E, Anderson A, Cohen-Gadol A, Hundley HA (2017) Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J Biol Chem 292:4326–4335. https://doi.org/10.1074/jbc.M117.779868
Okubo M, Horinishi A, Saito M et al (2007) A novel complex deletion–insertion mutation mediated by Alu repetitive elements leads to lipoprotein lipase deficiency. Mol Genet Metab 92:229–233. https://doi.org/10.1016/J.YMGME.2007.06.018
Oliver KR, Greene WK (2011) Mobile DNA and the TE-Thrust hypothesis: supporting evidence from the primates. Mob DNA 2:8. https://doi.org/10.1186/1759-8753-2-8
Ottesen EW, Seo J, Singh NN, Singh RN (2017) A multilayered control of the human survival motor neuron gene expression by Alu elements. Front Microbiol 8:2252. https://doi.org/10.3389/fmicb.2017.02252
Pal S, Tyler JK (2016) Epigenetics and aging. Sci Adv 2:e1600584–e1600584. https://doi.org/10.1126/sciadv.1600584
Pandolfo M (2006) Friedreich ataxia: detection of GAA repeat expansions and frataxin point mutations. In: Congenital Heart Disease. Humana Press, New Jersey, pp 197–216
Paquola ACM, Erwin JA, Gage FH (2016) Insights into the role of somatic mosaicism in the brain. Curr Opin Syst Biol 1:90–94
Payer LM, Steranka JP, Yang WR et al (2017) Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc Natl Acad Sci USA 114:E3984–E3992. https://doi.org/10.1073/pnas.1704117114
Payton A, Sindrewicz P, Pessoa V et al (2016) A TOMM40 poly-T variant modulates gene expression and is associated with vocabulary ability and decline in non-pathological aging. Neurobiol Aging 39:217 e1-7
Pehlivan D, Beck CR, Okamoto Y et al (2016) The role of combined SNV and CNV burden in patients with distal symmetric polyneuropathy. Genet Med 18:443–451. https://doi.org/10.1038/gim.2015.124
Petschner P, Gonda X, Baksa D et al (2017) Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience. https://doi.org/10.1016/J.NEUROSCIENCE.2017.09.049
Picardi E, Manzari C, Mastropasqua F et al (2015) Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep 5:14941. https://doi.org/10.1038/srep14941
Picardi E, D’Erchia AM, Lo Giudice C, Pesole G (2017a) REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res 45:D750–D757. https://doi.org/10.1093/nar/gkw767
Picardi E, Horner DS, Pesole G (2017b) Single-cell transcriptomics reveals specific RNA editing signatures in the human brain. RNA 23:860–865. https://doi.org/10.1261/rna.058271.116
Polak P, Domany E (2006) Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics 7:133. https://doi.org/10.1186/1471-2164-7-133
Prendergast JGD, Chambers EV, Semple CAM (2014) Sequence-level mechanisms of human epigenome evolution. Genome Biol Evol 6:1758–1771. https://doi.org/10.1093/gbe/evu142
Prudencio M, Gonzales PK, Cook CN et al (2017) Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet 26:3421–3431. https://doi.org/10.1093/hmg/ddx233
Prüfer K, Munch K, Hellmann I, et al (2012) The bonobo genome compared with the chimpanzee and human genomes Nature 486:nature11128. doi: https://doi.org/10.1038/nature11128
Reddy PH (2011) Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 1415:136–148
Reynolds WF, Rhees J, Maciejewski D et al (1999) Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp Neurol 155:31–41. https://doi.org/10.1006/exnr.1998.6977
Rezin GT, Amboni G, Zugno AI et al (2009) Mitochondrial dysfunction and psychiatric disorders. Neurochem Res 34:1021–1029. https://doi.org/10.1007/s11064-008-9865-8
Rice GI, Kasher PR, Forte GMA et al (2012) Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet 44:1243–1248. https://doi.org/10.1038/ng.2414
Roger AJ, Muñoz-Gómez SA, Kamikawa R (2017) The origin and diversification of mitochondria. Curr Biol 27:R1177–R1192. https://doi.org/10.1016/j.cub.2017.09.015
Roses AD (2010) An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Arch Neurol 67:536–541
Roses AD, Lutz MW, Amrine-Madsen H et al (2010) A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J 10:375–384
Roses AD, Sundseth S, Saunders A et al (2016a) Understanding the genetics of APOE and TOMM40 and role of mitochondrial structure and function in clinical pharmacology of Alzheimer’s disease. Alzheimers Dement 12:687–694. https://doi.org/10.1016/j.jalz.2016.03.015
Roses AD, Akkari PA, Chiba-Falek O et al (2016b) Structural variants can be more informative for disease diagnostics, prognostics and translation than current SNP mapping and exon sequencing. Expert Opin Drug Metab Toxicol 12:135–147. https://doi.org/10.1517/17425255.2016.1133586
Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med:S10–S17
Rossi L, Lombardo MF, Ciriolo MR, Rotilio G (2004) Mitochondrial dysfunction in neurodegenerative diseases associated with copper imbalance. Neurochem. Res. 29:493–504
Rugarli EI, Langer T (2012) Mitochondrial quality control: a matter of life and death for neurons. EMBO J 31:1336–1349
Rybak-Wolf A, Stottmeister C, Glazar P et al (2015) Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell 58:870–885
Sakurai M, Ueda H, Yano T et al (2014) A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res 24:522–534. https://doi.org/10.1101/gr.162537.113
Saunders AM, Schmader K, Breitner JC et al (1993) Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer’s disease and in other amyloid-forming diseases. Lancet (London, England) 342:710–711. https://doi.org/10.1016/0140-6736(93)91709-U
Schanze D, Neubauer D, Cormier-Daire V et al (2014) Deletions in the 3′ part of the NFIX gene including a recurrent Alu-mediated deletion of exon 6 and 7 account for previously unexplained cases of Marshall-Smith syndrome. Hum Mutat 35:1092–1100. https://doi.org/10.1002/humu.22603
Schmauss C, Howe JR (2002) RNA editing of neurotransmitter receptors in the mammalian brain. Sci STKE 2002:pe26. https://doi.org/10.1126/stke.2002.133.pe26
Schneider I, Kugel H, Redlich R et al (2017) Association of serotonin transporter gene AluJb methylation with major depression, amygdala responsiveness, 5-HTTLPR/rs25531 polymorphism, and stress. Neuropsychopharmacology online early:1–9. https://doi.org/10.1038/npp.2017.273
Schollen E, Keldermans L, Foulquier F et al (2007) Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Mol Genet Metab 90:408–413. https://doi.org/10.1016/j.ymgme.2007.01.003
Schwahn U, Lenzner S, Dong J et al (1998) Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet 19:327–332. https://doi.org/10.1038/1214
Schwarz TL (2013) Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5:a011304
Seeley WW (2017) Mapping neurodegenerative disease onset and progression. Cold Spring Harb Perspect Biol 9:a023622. https://doi.org/10.1101/cshperspect.a023622
Selkoe DJ (2000) Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci 924:17–25. https://doi.org/10.1111/j.1749-6632.2000.tb05554.x
Shen S, Lin L, Cai JJ et al (2011) Widespread establishment and regulatory impact of Alu exons in human genes. Proc Natl Acad Sci 108:2837–2842
Shiota T, Imai K, Qiu J et al (2015) Molecular architecture of the active mitochondrial protein gate. Science 349:1544–1548
Shpyleva S, Melnyk S, Pavliv O et al (2017) Overexpression of LINE-1 retrotransposons in autism brain. Mol Neurobiol:1–10. https://doi.org/10.1007/s12035-017-0421-x
Skovronsky DM, Doms RW, Lee VM (1998) Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol 141:1031–1039
Smalheiser NR (2014) The RNA-centred view of the synapse: non-coding RNAs and synaptic plasticity. Philos Trans R Soc B Biol Sci 369:20130504–20130504. https://doi.org/10.1098/rstb.2013.0504
Soibam B (2017) Super-lncRNAs: identification of lncRNAs that target super-enhancers via RNA:DNA:DNA triplex formation. RNA 23:1729–1742
Steen E, Terry BM, Rivera EJ et al (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J Alzheimers Dis 7:63–80
Strittmatter WJ, Saunders AM, Schmechel D et al (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci 90:1977–1981. https://doi.org/10.1073/pnas.90.5.1977
Sun Z, Wu Y, Ordog T et al (2014) Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E. Epigenetics 9:1184–1193. https://doi.org/10.4161/epi.29676
Swerdlow RH (2017) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimer’s Dis Preprint:1–14. https://doi.org/10.3233/JAD-170585
Swerdlow RH, Burns JM, Khan SM (2010) The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimer’s Dis JAD 20:265
Szpakowski S, Sun X, Lage JM et al (2009) Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene 448:151–167. https://doi.org/10.1016/j.gene.2009.08.006
Takahashi RH, Milner TA, Li F et al (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161:1869–1879
Tan B, Huang H, Lam R, Soong T (2009) Dynamic regulation of RNA editing of ion channels and receptors in the mammalian nervous system. Mol Brain 2:13. https://doi.org/10.1186/1756-6606-2-13
Tan MH, Li Q, Shanmugam R et al (2017) Dynamic landscape and regulation of RNA editing in mammals. Nature 550:249–254. https://doi.org/10.1038/nature24041
Tarallo V, Hirano Y, Gelfand BD et al (2012) DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149:847–859. https://doi.org/10.1016/j.cell.2012.03.036
Tariq A, Jantsch MF (2012) Transcript diversification in the nervous system: A to I RNA editing in CNS function and disease development. Front Neurosci 1–13. https://doi.org/10.3389/fnins.2012.00099
Taşkesen M, Collin GB, Evsikov AV et al (2012) Novel Alu retrotransposon insertion leading to Alström syndrome. Hum Genet 131:407–413. https://doi.org/10.1007/s00439-011-1083-9
Thongsroy J, Patchsung M, Mutirangura A (2017) The association between Alu hypomethylation and severity of type 2 diabetes mellitus. Clin Epigenetics 9:93. https://doi.org/10.1186/s13148-017-0395-6
Töhönen V, Katayama S, Vesterlund L et al (2015) Novel PRD-like homeodomain transcription factors and retrotransposon elements in early human development. Nat Commun 6:8207. https://doi.org/10.1038/ncomms9207
Tsirigos A, Rigoutsos I (2009) Alu and B1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLOS Comput Biol 5:e1000610. https://doi.org/10.1371/JOURNAL.PCBI.1000610
Udaka T, Okamoto N, Aramaki M et al (2007) An Alu retrotransposition-mediated deletion of CHD7 in a patient with CHARGE syndrome. Am J Med Genet Part A 143:721–726. https://doi.org/10.1002/ajmg.a.31441
Ullu E, Tschudi C (1984) Alu sequences are processed 7SL RNA genes. Nature 312:171–172. https://doi.org/10.1038/312171a0
Van Den Heuvel MP, Sporns O (2013) Network hubs in the human brain. Trends Cogn Sci 17:683–696. https://doi.org/10.1016/j.tics.2013.09.012
Vandewalle J, Bauters M, Van Esch H et al (2013) The mitochondrial solute carrier SLC25A5 at Xq24 is a novel candidate gene for non-syndromic intellectual disabilty. Hum Genet 132:1177–1185
Vansant G, Reynolds WF (1995) The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc Natl Acad Sci U S A 92:8229–8233
Varizhuk AM, Sekridova AV, Tankevich MV et al (2016) Conformational polymorphysm of G-rich fragments of DNA Alu-repeats. II. the putative role of G-quadruplex structures in genomic rearrangements. Biomeditsinskaya Khimiya 62:630–637. https://doi.org/10.18097/pbmc20166206630
Varshney D, Vavrova-Anderson J, Oler AJ, et al (2015) SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation.
Vassetzky NS, Ten OA, Kramerov DA (2003) B1 and related SINEs in mammalian genomes. Gene 319:149–160
Viader A, Golden JP, Baloh RH et al (2011) Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J Neurosci 31:10128–10140. https://doi.org/10.1523/JNEUROSCI.0884-11.2011
Viader A, Sasaki Y, Kim S et al (2013) Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 77:886–898. https://doi.org/10.1016/j.neuron.2013.01.012
Wang W, Li L, Lin W et al (2013) The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet 22:4706–4719
Wang W, Wang WH, Azadzoi KM et al (2016) Alu RNA accumulation in hyperglycemia augments oxidative stress and impairs eNOS and SOD2 expression in endothelial cells. Mol Cell Endrocrinol 426:91–100
Weissmann D, van der Laan S, Underwood MD et al (2016) Region-specific alterations of A-to-I RNA editing of serotonin 2c receptor in the cortex of suicides with major depression. Transl Psychiatry 6:e878. https://doi.org/10.1038/tp.2016.121
Wimmer K, Callens T, Wernstedt A, Messiaen L (2011) The NF1 gene contains hotspots for L1 endonuclease-dependent De Novo insertion. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002371
Wright A, Vissel B (2012) The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front Mol Neurosci 5:34. https://doi.org/10.3389/fnmol.2012.00034
Wu S-J, Hsieh T-J, Kuo M-C, et al (2013) Functional regulation of Alu element of human angiotensin-converting enzyme gene in neuron cells. Neurobiol Aging 34:1921.e1-1921921.e7. doi: https://doi.org/10.1016/j.neurobiolaging.2013.01.003
Zempel H, Mandelkow E (2015) Tau missorting and spastin-induced microtubule disruption in neurodegeneration: Alzheimer disease and hereditary spastic paraplegia. Mol Neurodegener 10:68. https://doi.org/10.1186/s13024-015-0064-1
Zhang Y-H, Dipple KM, Vilain E et al (2000) AluY insertion (IVS4-52ins316alu) in the glycerol kinase gene from an individual with benign glycerol kinase deficiency. Hum Mutat 15:316–323. https://doi.org/10.1002/(SICI)1098-1004(200004)15:4<316::AID-HUMU3>3.0.CO;2-9
Zhang G, Fukao T, Sakurai S et al (2006) Identification of Alu-mediated, large deletion-spanning exons 2-4 in a patient with mitochondrial acetoacetyl-CoA thiolase deficiency. Mol Genet Metab 89:222–226. https://doi.org/10.1016/j.ymgme.2006.06.010
Acknowledgements
We thank A.D. Brown, W.K. Gottschalk, and M. Mihovilovic for helpful discussion. C. Hohoff, B. Ciapa, and an anonymous reviewer kindly reviewed the manuscript and provided helpful comments. This is Duke Lemur Center publication number 1389. We dedicate this work to the memory of Allen Roses, who believed there was no such thing as “junk DNA”.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Beth Sullivan.
Electronic supplementary material
ESM 1
(XLSX 31 kb).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Larsen, P.A., Hunnicutt, K.E., Larsen, R.J. et al. Warning SINEs: Alu elements, evolution of the human brain, and the spectrum of neurological disease. Chromosome Res 26, 93–111 (2018). https://doi.org/10.1007/s10577-018-9573-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-018-9573-4