
Abstract
We review the picture of chromatin large-scale 3D organization emerging from the analysis of Hi-C data and polymer modeling. In higher mammals, Hi-C contact maps reveal a complex higher-order organization, extending from the sub-Mb to chromosomal scales, hierarchically folded in a structure of domains-within-domains (metaTADs). The domain folding hierarchy is partially conserved throughout differentiation, and deeply correlated to epigenomic features. Rearrangements in the metaTAD topology relate to gene expression modifications: in particular, in neuronal differentiation models, topologically associated domains (TADs) tend to have coherent expression changes within architecturally conserved metaTAD niches. To identify the nature of architectural domains and their molecular determinants within a principled approach, we discuss models based on polymer physics. We show that basic concepts of interacting polymer physics explain chromatin spatial organization across chromosomal scales and cell types. The 3D structure of genomic loci can be derived with high accuracy and its molecular determinants identified by crossing information with epigenomic databases. In particular, we illustrate the case of the Sox9 locus, linked to human congenital disorders. The model in-silico predictions on the effects of genomic rearrangements are confirmed by available 5C data. That can help establishing new diagnostic tools for diseases linked to chromatin mis-folding, such as congenital disorders and cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The traditional view of DNA as a linear sequence of nucleotides carrying genetic information has radically changed over the last couple of decades as its spatial organization inside the cell nucleus has been shown to play a key role in gene regulation, in normal cells as well as in diseases (Bickmore and van Steensel 2013; Lanctôt et al. 2007; Misteli 2007; Tanay and Cavalli 2013; Dekker and Mirny 2016). New technologies, such as Hi-C (Lieberman-Aiden et al. 2009), are revealing complex 3D patterns of genomic contacts in the mammalian genome and their analysis is returning a formidable picture of chromosome folding (Nora et al. 2012; Dixon et al. 2012; Phillips-Cremins et al. 2013; Fraser et al. 2015). Here, we summarize recent discoveries about the large-scale architecture of chromosomes during neuronal differentiation. Recent studies have shown that chromatin is folded in a tree-like hierarchy of domains-within-domains, or metaTADs, extending across genomic scales (Fraser et al. 2015). Far from random, contacts between regions with similar functional properties, for example active genes, tend to have preferential co-localization, and occupy common structural domains correlated to the structure of metaTADs. Such a hierarchical organization may reflect the capacity for general compaction of the genome while retaining the flexibility needed for regional specialization of nuclear functions.
To understand the molecular mechanisms whereby chromosome 3D structure is shaped and orchestrated in the cell nucleus, concepts from polymer physics have been introduced in order to provide a quantitative, predictive framework to explain experimental contact patterns in a principled approach (see, e.g., a review in Nicodemi and Pombo 2014; and Nicodemi and Prisco 2009; Bohn and Heermann 2010; Barbieri et al. 2012; Brackley et al. 2013; Jost et al. 2014; Sanborn et al. 2015; Brackley et al. 2016a, b; Fudenberg et al. 2016; Chiariello et al. 2016; Di Stefano et al. 2016). Here, in particular, we discuss that chromatin has been shown to be a complex mixture of different regions, folded in the classical architectural classes predicted by polymer thermodynamics (pure states) (Nicodemi and Prisco 2009; Barbieri et al. 2012). Hi-C contact matrices can be derived genome-wide with high accuracy and their molecular determinants identified by the theory (Chiariello et al. 2016). In particular, we describe the case of the Sox9 locus, a region linked to severe human congenital diseases. In mESC, that locus self-assembles hierarchically in higher-order domains, involving abundant many-body contacts. The model predictions on the effects of structural rearrangements are tested in the case of the ΔXTX deletion in the Xist locus, where 5C data are available in wild-type and mutant cells.
Higher-order chromatin folding
By the analysis of Hi-C interactions, it has been discovered that chromosomes are divided in 0.5-1 Mb long domains, known as topologically associated domains (TADs) (Dixon et al. 2012; Nora et al. 2012), heuristically defined as regions marked by increased levels of intra-domain Hi-C interactions. Larger structures have been also identified, such as the 10 Mb wide A/B compartments linked respec-tively to more transcribed and repressed chromatin sequences (Lieberman-Aiden et al. 2009). Yet, patterns are visible also within and across TADS at lower as much as at very large scales (Phillips-Cremins et al. 2013).
Higher-order chromatin 3D structures were identified by analyzing the interactions between TADs, through mouse neuronal differentiation. Chromatin was found to be organized in a hierarchy of domains-within-domains, named metaTADs, up to chromosomal scales (Fraser et al. 2015). The heuristic method to define such metaTADs (Fig. 1) was based on the consideration that the best candidates to form higher-order architectural structures are the pairs of most strongly interacting TADs, as by definition they share a number of contacts: hence, the most strongly interacting pair of domains is clustered to form a metaTAD and the procedure repeated iteratively (Fig. 1 left). In this way, a tree of metaTADs is produced that represents the way the different TADs aggregate to form higher-order structures.

Chromatin in higher mammals is organized in a hierarchy of domains-within-domains. a) The heatmap shows the Hi-C contact map of a 5 Mb region of chromosome 2 (53-58 Mb) in mESC (Fraser et al. 2015). Topologically Associating Domains (TADs) are identified via the Directionality Index (DI (Dixon et al. 2012; Fraser et al. 2015)) and correspond to the squares along the diagonal marked by black numbers. (named 1–6). b) TADs contact each other in groups forming higher-order domains, named metaTADs (marked by roman numbers), identified by iteratively clustering the most strongly Hi-C interacting domain pair. c) MetaTADs have been shown to be statistically significant architectural structures up to chromosomal scales, validated by FISH (Fraser et al. 2015). Those analyses return a picture of chromosomes where they are folded in a complex system of higher-order structures, organized as a hierarchy of domains-within-domains. (Adapted from Fraser et al. 2015)
To validate the architectural relevance of such metaTADs from a statistical viewpoint different measures were employed (Fraser et al. 2015). As much as for the more fundamental TADs, it has been shown, for example, that Hi-C interactions within and across metaTADs were significantly above background up to chromosomal scales. They were also confirmed by FISH. MetaTADs are, thus, relevant architectural structures, as much as TADs.
To assess the biological relevance of metaTADs, the enrichment of chromatin features, known to mark TAD boundaries, was explored at metaTAD boundaries to discover that they are even more enriched (Fig. 2). More generally, the hierarchical 3D architecture of metaTADs itself was shown to correlate with a variety of epigenomic signals (Fraser et al. 2015). Taken together such results point out that metaTADs must be at least as significant as the fundamental TADs they are composed of.

MetaTAD boundaries are enriched with chromatin features. TAD boundaries are known to be enriched by a variety of epigenetic features, a notable example being CTCF binding sites (Nora et al. 2012; Dixon et al. 2012). Interestingly metaTAD boundaries are even more enriched for the same factors (Fraser et al. 2015), highlighting than they are as biological significant. More generally, the hierarchical 3D architecture of metaTADs itself correlates with epigenomics, beyond their boundaries. (Adapted from Fraser et al. 2015)
The hierarchical organization of chromatin in domains-within-domains is systematically found during neuronal differentiation, and it is confirmed consistently in other mouse and human cell types (Fraser et al. 2015). The metaTAD trees of chromosomes are, though, only relatively conserved during differentiation, as their average correlation across time points in approximately 82% (cophenetic correlation, Fig. 3). This value is comparable to the variation of TAD boundaries that are approximately 70% conserved in the transitions from mESC to neural precursors (NPC) and post-mitotic neurons (Fraser et al. 2015), a figure similar to recent findings in other systems (Schmitt et al. 2016). Architectural changes are subtly linked to variations in transcriptional activity: expression changes overlap with both changing and conserved metaTAD tree regions, but tend to be coherent in sign only in the latter. Stated differently, the analysis of CAGE transcription data, matched with Hi-C data, has revealed that TADs belonging to the same conserved niche of the metaTAD tree tend to change their transcriptional activity in the same direction. Interestingly, it has been also reported that expression changes overlap mostly with compartment A or A-B transitions, while conserved expression regions mostly with B (Fraser et al. 2015).

Conformational reorganization of metaTADs upon neuronal differentiation in mouse models. MetaTAD architecture is only relatively conserved during neuronal differentiation in mouse models (average Cophenetic correlation approx. 80%) (Fraser et al. 2015). The figure compares the metaTAD tree of chr6 in mESC and neural precursor cells (NPC). The degree of local changes in the tree structure is shown by the central color map. TADs in architecturally conserved niches tend to have expression changes coherent in sign. (Adapted from Fraser et al. 2015)
Taken together the above results return a picture where chromatin architecture is organized in a hierarchy of domains-within-domains (metaTADs), which correlates with genetic, epigenetic and transcription data (see scheme in Fig. 1c). The different levels of metaTADs encompass the scales of TADs (0.5 Mb) and of A/B compartments (10 Mb) up to chromosomal levels, in mouse and human cell types.
Polymer models of chromosomes
The above definitions of domains (such as TADs, A/B compartments and metaTADs) are heuristic, derived by simple criteria to empirically identify patterns in Hi-C data. In the last few years, principled approaches have been introduced to try to identify the nature of chromatin architectural domains and their molecular determinants from the basic laws of polymer physics (Nicodemi and Pombo 2014).
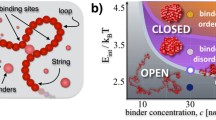
An important element in chromatin folding has been shown to be steric hindrance effects (Kreth et al. 2004; Marenduzzo et al. 2006; Rosa and Everaers 2008; Lieberman-Aiden et al. 2009), however polymer models that also take into account interactions with diffusing molecular factors are needed to describe FISH and Hi-C data in a more quantitative way (Nicodemi and Prisco 2009; Bohn and Heermann 2010; Barbieri et al. 2012; Brackley et al. 2013; Jost et al. 2014; Sanborn et al. 2015; Brackley et al. 2016a, b; Fudenberg et al. 2016; Chiariello et al. 2016; Di Stefano et al. 2016). To describe in quantitative terms the scenario where chromosome conformations are shaped by interactions with nuclear elements such as DNA-binding molecules, here we consider the Strings&Binders (SBS) polymer model (Nicodemi and Prisco 2009) that has been shown to well recapitulate Hi-C, as well as FISH data within a single framework (Barbieri et al. 2012; Fraser et al. 2015) (Fig. 4a). In such a model a chromatin filament is represented as a SAW (Self-Avoiding Walk) polymer chain with binding sites for diffusing binders. In a toy version of the model (Fig. 4a) only one type of binding sites is present: the concentration of binders is named c and their affinity E X . The system is subject to the laws of physics and let to fold in its stable conformational states where loops are spontaneously established by the attachment of the binders at their cognate binding sites along the chain.

A cartoon of the Strings&Binders polymer model and its phase diagram. a) A cartoon of a simple version of the Strings&Binders (SBS) model including only one type of binders (red spheres): c is the binder concentration and E Int their binding affinity. Its equilibrium conformations fall into universality classes that are in a one to one correspondence with the system thermodynamics phases. b) The phase diagram of the model in panel a) includes (Chiariello et al. 2016): a coil phase where the chain is open, a Θ-point transition line and a globule phase where it is closed in compact conformations. In the globule phase, the binders have also an order-disorder transition that can lead to ordered structures. (Adapted from Chiariello et al. 2016)
From polymer physics it is known that the SBS polymer equilibrium conformations fall into a few universality classes that are in a one to one correspondence with its thermodynamics phases. Different conformations of the polymer belonging to the same class are statistically indistinguishable. Hence, to derive the polymer folding properties it is enough to identify the system thermodynamics phases. Figure 4b reports the phase diagram of the toy model in Fig. 4a where only one type of binding sites and binders is present (red) (Chiariello et al. 2016). The system has three main phases: at low concentrations, c, or small affinities, E X , the chain is in its coil state, randomly folded as on open chain; above its Θ-point transition, a globule phase exists where the chain is closed in compact conformations, while the binders can form a disordered or an ordered (crystalline-like) assembly according to the value of c and E X .
The conformational classes identified by the coil-globule and order-disorder transitions are general features of the physics of interacting polymers, independent on finer model details. Although in real chromatin off-equilibrium conformations are also expected to be encountered, those thermodynamics classes represent the stable states where the system spontaneously folds into according to the laws of physics and, as such, they must be an important component in chromosome folding, at least at the level of single loci. In fact, eu- and heterochromatin domains, visualized as open and compact states, are well known in microscopy. Hence, a natural, yet approximated physics model of chromatin is to consider a chromosome as a sequence of regions folded in the architectural classes predicted by polymer physics, such as open and compact conformations (Barbieri et al. 2012; Chiariello et al. 2016).
A more realistic model of chromatin architectural domains
Interestingly, within such a simplified view based on fundamental polymer physics, a more realistic version of the SBS polymer model can explain Hi-C pairwise contact data with good accuracy. For instance, in the case of a 6 Mb region around the Sox9 gene (important in human congenital diseases) in mESC (Fig. 5a), the patterns of Hi-C data is reproduced with a 95% accuracy by the model as seen in Fig. 5b (Chiariello et al. 2016). This is achieved by a more refined SBS polymer with respect to the toy model of Fig. 4a, including 15 different types of binders and cognate binding sites (see cartoon in Fig. 5a), whose genomic locations are shown in Fig. 5c represented by different colors. The position along the sequence of the binding domains (colors) is derived from the experimental Hi-C contact data, via a robust procedure of statistical inference, in order to obtain the best, minimal SBS polymer model explaining such data based only on physics (Chiariello et al. 2016).

The SBS model can explain folding of the Sox9 locus with good accuracy. a) The 6 Mb wide Sox9 locus in mESC is shown along with a cartoon of its SBS polymer model. b) The Hi-C pairwise contact data of the region (top) are explained with a 95% Pearson correlation by its SBS model (contact matrix at the bottom) (Chiariello et al. 2016). c) The SBS model of the locus predicts 15 different binding domains. We developed an approach where the binding factors as much as their genomic location are derived by only architectural data, with no need of previous knowledge of DNA binding molecules. The genomic positions of the binding domains of Sox9, represented by different colors, are here shown along with the TADs of the locus. The predicted binding domains overlap with each other, producing the complex patterns visible within TADs and TAD-TAD interactions into higher-order metaTAD structures. (Adapted from Chiariello et al. 2016)
The body of the different binding domains is roughly coinciding with the known different TADs of the locus (A-F in Fig. 5c). Yet, binding domains overlap with each other, which explains the complex internal structures seen within TADs and at TAD borders. Some of the domains extend broadly over the region to produce the interactions across TADs seen in Hi-C data, which give rise to the higher-order structures described by metaTADs (Chiariello et al. 2016). In this picture, the binding domains are the physical, fundamental determinants of chromatin architectural patterns, heuristically visualized as TADs and metaTADs. As the SBS model can explain comparatively well Hi-C data, it can provide a mechanistic understanding of the regulation of chromosome architecture, capturing at least some of their key folding mechanisms.
Importantly, our polymer models can be used to derive any information on the folding of the loci of interest genome-wide, beyond Hi-C pairwise contact data. For instance, a snapshot of the full 3D structure of the Sox9 locus, along with a comparison with its average contact matrix and TADs, is shown in Fig. 6a. For example, it is visible that TAD D (red) has a complex internal 3D structure, with a large part of it mostly associated to TAD E. Additionally, the dynamics of the interactions between the genes within the locus and their regulators can be derived (Chiariello et al. 2016). An important, general discovery based on such polymer models is that many-body interactions (triplets, quadruplets, etc.) are much more abundant than expected in a randomly folded polymer chain (Fig. 6b), highlighting that they could be an important functional component in chromatin regulation.

The 3D structure of the Sox9 locus derived by the SBS model and its molecular determinants. a) A snapshot of the full 3D structure of the Sox9 locus in mESC derived by its SBS model. It allows to rationalizing the patterns seen in its Hi-C map and the relative organization and interactions of TADs. b) Beyond the pairwise interactions seen by Hi-C, the model predicted 3D structure reveal many-body contacts much more abundant than in a randomly folded chain, highlighting a likely functional role in chromatin regulation (Chiariello et al. 2016). c) Heatmap of the correlations between the genomic positions of the binding domains in the Sox9 SBS model and chromatin features. A barcode links architectural domains to epigenomic marks, pointing towards a combinatorial regulation of chromatin folding and transcriptional activities
Molecular nature of the polymer binding domains
To infer the specific molecular nature of the predicted binding sites (model colors) and their cognate binding factors, we crossed the information on their position/type with epigenomic databases of chromatin features. By integration of the data we can identify known and new candidate factors driving folding and responsible for its regulation. The heatmap in Fig. 6c illustrates the correlation coefficient between the genomic positions of binding domains and chromatin features from ENCODE (The ENCODE Project Consortium 2012) in the Sox9 region in mESC. Each binding domain appears to have an epigenetic barcode that is a unique combinatorial pattern of epigenetic features. For instance, some domains are characterized by active marks and PolII, others by more repressive marks. CTCF correlates strongly with many of the domains, yet others are not linked to it. That points out that additional architectural factors, beyond CTCF, play a key role in folding.
A test of the model
A stringent test of the model can be made by comparing its predictions on the effects on folding of structural variants, against their corresponding Hi-C patterns in cells bearing the mutations. Starting from wild-type data, the SBS model of any locus can be derived as seen in the Sox9 case above. The structural variant of interest is then implemented into the model and its new 3D conformation derived under only the laws of physics, without any adjustable parameter whatsoever. The resulting pairwise contact data can then be compared with Hi-C data of the modified cell system. We tested the model prediction in the case of the Xist locus (Fig. 7a) where 5C data are available for the wild-type locus and for the ΔXTX deletion in mESC (Nora et al. 2012; Giorgietti et al. 2014). Interestingly, the model well reproduces (90% correlation) the observed ectopic interactions emerging as a consequence of ΔXTX in 5C data (Fig. 7b,c).

Test of the SBS model: the effects of deletions on the 3D architecture of the Xist locus can be predicted. a) For the shown 1 Mb wide Xist locus, 5C data are available in wild-type mESC and in the case of cells bearing the ΔXTX deletion (Nora et al. 2012). b) The contact matrix (bottom) of the SBS model best describing the wild-type locus has a 96% Pearson correlation with 5C experimental data (top) (Chiariello et al. 2016). From the inferred polymer model, the effects of ΔXTX on folding can be predicted from the laws of physics under no adjustable parameters, providing a stringent test against experimental data. c) Interes-tingly, the predicted contact matrix has a 91% correlation with 5C data of the deletion and identifies the ectopic contacts arising after the deletion. d) Additionally, from the SBS model the full 3D structure can be derived, beyond the available pairwise contact data, to visualize the effects of the mutation on folding (Chiariello et al. 2016). (Adapted from Chiariello et al. 2016)
The full 3D structure of the wild-type and of the deleted system are shown in the snapshots of Fig. 7d (Chiariello et al. 2016), highlighting that a part of the ectopic interactions seen in ΔXTX derive from mere proximity effects.
In summary, by suitable polymer models and by crossing information with epigenomic databases we can develop a platform to dissect the molecular factors controlling folding and defining chromatin domains in a principled way. As a consequence, we can also predict the effects of the 3D organization of chromosomes of structural variants, along with the rewiring of the regulatory networks of genes and their regulators. As architecture and transcription are deeply linked, that can open the way to understand disease models, such as congenital diseases and cancer.
Discussion
The approach based on the SBS model discussed above has been used to explain folding at other loci, such as the Bmp7 and the HoxB regions in mESC, along with their dynamics (Chiariello et al. 2016; Annunziatella et al. 2016). Polymer models, in the same category than the SBS model, informed with protein binding sites (i.e., DHS maps and CTCF sites) have also successfully reproduced the pattern of contacts at loci such as the α and β globin gene regions in mouse erythroblasts (Brackley et al. 2016a, b), the Xist locus in mouse embryonic stem cells (Scialdone et al. 2011, Giorgietti et al. 2014; Chiariello et al. 2016) or genomic loci in Drosophila (Jost et al. 2014) and budding yeast (Cheng et al. 2015). Polymer modeling of chromatin structural organization has also been linked to replication timing domains (Boulos et al. 2015). Finally, models based on an active, DNA loop extruding mechanisms have been shown to be compatible with Hi-C contact data at loci where folding is driven by CTCF binding sites (Sanborn et al. 2015; Fudenberg et al. 2016). However, for instance, the 3D structures of the globin loci have been shown to remain preserved in CTCF or other Transcription-Factor knock-outs (Brackley et al. 2016a, b). Analogously, in the mouse Sox9 locus, CRISPR/Cas9 precise deletions of TAD boundary regions containing conserved CTCF binding sites had no special effects on Hi-C contact maps and produced no alterations of TADs (Franke et al. 2016). Such results point towards a scenario where the combination of different molecular factors is responsible for chromatin folding, as predicted by the SBS model discussed here.
In conclusion, we discussed recent developments based on the analysis of Hi-C data genome-wide, in human as well as mouse cell models, that reveal that chromosomes have a complex hierarchical architecture of higher-order domains (metaTADs) extending up to chromosomal scales, in a manner that reflects epigenomic features. MetaTAD topologies are relatively conserved through differentiation and their reorganization is related to gene expression changes. We also reviewed how concepts of polymer physics can explain chromatin 3D structures from the sub-Mbp to chromosomal scales, across cell types and chromosomes. In particular, we discussed recent findings showing that the 3D structure of loci, such as the Sox9 locus, linked to human congenital disorders, can be explained with high accuracy and its molecular determinants identified. Model predictions on the effects of genomic rearrangements have been confirmed against available data, e.g., in the Xist locus. Importantly, the approach proposed here does not require previous knowledge of DNA binding proteins. In our approach, the molecular factors shaping folding, as much as their genomic locations, are derived by only architectural data. Their molecular nature is later identified by crossing their positional information with epigenomics datasets. In this way, we can identify the determinants of folding and their mechanisms of actions with no a-priori assumptions beyond that chromosome conformations reflects polymer physics. Our approach can, thus, help developing new diagnostic and treatment tools for diseases linked to chromatin mis-folding, such as congenital disorders and cancer (Ong and Corces 2014; Lupiáñez et al. 2015; Valton and Dekker 2016).
Materials and methods
A detailed description of the materials and methods is provided in the cited references. In particular, the SBS polymer model was investigated by Brownian Molecular Dynamics computer simulations imple-mented by LAMMPS. All the details about the model and computational parameters, as well as on the analyses performed are reported in the cited references.
The data files of chromatin features in the Sox9 region in mESC used here are available online from ENCODE, accession numbers ENCFF434DJA, ENCFF916EVG, ENCFF945LRL, ENCFF001YIX, ENCFF001YAD, ENCFF001YHE, ENCFF817CZF, ENCFF001XWX, ENCFF001YIY, ENCFF001YIV, ENCFF830QDG, ENCFF001YAE, ENCFF883ROD, ENCFF530LPO, ENCFF854IVF and from GEO, accession numbers GSM1000126, GSM1000124.
Abbreviations
- CTCF:
-
CCCTC-binding factor
- FISH:
-
Fluorescence in situ hybridization
- mESC:
-
mouse embryonic stem cell
- NPC:
-
Neural precursor cell
- SBS:
-
Strings & Binders
- TAD:
-
Topologically associated domain
References
Annunziatella C, Chiariello AM, Bianco S, Nicodemi M (2016) Polymer models of the hierarchical folding of the Hox-B chromosomal locus. Phys Rev E 94:042402
Barbieri M, Chotalia M, Fraser J, Lavitas LM, Dostie J, Pombo A, Nicodemi M (2012) Complexity of chromatin folding is captured by the strings and binders switch model. Proc Natl Acad Sci U S A 109:16173–16178
Bickmore WA, van Steensel B (2013) Genome architecture: domain organization of interphase chromosomes. Cell 152:1270–1284
Bohn M, Heermann DW (2010) Diffusion-driven looping provides a consistent framework for chromatin organisation. PLoS One 5:e12218
Boulos RE, Drillon G, Argoul F, Arneodo A, Audit B (2015) Structural organization of human replication timing domains. FEBS Lett 589:2944–2957
Brackley CA, Brown JM, Waithe D et al (2016b) Predicting the three-dimensional folding of cis-regulatory regions in mammalian genomes using bioinformatic data and polymer models. Genome Biol 17:59
Brackley CA, Taylor S, Papantonis A, Cook PR, Marenduzzo D (2013) Nonspecific bridging-induced attraction drives clustering of DNA-binding proteins and and genome organisation. Proc Natl Acad Sci U S A 110:E3605–E3611
Brackley CA, Johnson J, Kelly S, Cook PR, Marenduzzo D (2016a) Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains. Nucl Acids Res 44:3503–3512
Cheng TMK, Heeger S, Chaleil RAG et al (2015) A simple biophysical model emulates budding yeast chromosome condensation. eLife 4:e05565
Chiariello AM, Annunziatella C, Bianco S, Esposito A, Nicodemi M (2016) Polymer physics of chromosome large-scale 3d organisation. Scientific Reports 6:29775
Dekker J, Mirny L (2016) 3D genome as moderator of chromosomal communication. Cell 164:1110–1121
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380
Di Stefano M, Paulsen J, Lien TG, Hovig E, Micheletti C (2016) Hi-C-constrained physical models of human chromosomes recover functionally-related properties of genome organization. Scientific Reports 6:35985
Franke M, Ibrahim DM et al (2016) Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538:265–269
Fraser J, Ferrai C, Chiariello AM et al (2015) Hierarchical folding and reorganisation of chromosomes are linked to transcriptional changes during cellular differentiation. Mol Syst Biol 11:852
Fudenberg G, Imakaev M, Lu C et al (2016) Formation of chromosomal domains by loop extrusion. Cell Rep 15:1–12
Giorgietti L, Galupa R, Nora EP et al (2014) Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell 157:950–963
Jost D, Carrivain P, Cavalli G, Vaillant C (2014) Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res 42:9553–9561
Kreth G, Finsterle J, von Hase J, Cremer M, Cremer C (2004) Radial arrangement of chromosome territories in human cell nuclei: a computer model approach based on gene density indicates a probabilistic global positioning code. Biophys J 86:2803–2812
Lanctôt C, Cheutin T, Cremer M, Cavalli G, Cremer T (2007) Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet 8:104–115
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293
Lupiáñez DG, Kraft K, Heinrich V et al (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161:1
Marenduzzo D, Micheletti C, Cook PR (2006) Entropy-driven genome organisation. Biophys J 90:3712–3721
Misteli T (2007) Beyond the sequence: cellular organization of genome function. Cell 128:787–800
Nicodemi M, Pombo A (2014) Models of chromosome structure. Curr Opin Cell Biol 28C:90–95
Nicodemi M, Prisco A (2009) Thermodynamic pathways to genome spatial organization in the cell nucleus. Biophys J 96:2168–2177
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J, Heard E (2012) Spatial partitioning of the regulatory landscape of the X-inactivation Centre. Nature 485:381–385
Ong CT, Corces VG (2014) CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15:234–246
Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG (2013) Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153:1281–1295
Rosa A, Everaers R (2008) Structure and dynamics of interphase chromosomes. PLoS Comput Biol 4:e1000153
Sanborn AL, Rao SSP, Huang S-C et al (2015) Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112:E6456–E6465
Scialdone A, Cataudella I, Barbieri M, Prisco A, Nicodemi M (2011) Conformation regulation of the X chromosome inactivation center: a model. PLoS Comput Biol 7:e1002229
Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, Li Y, Lin S, Lin Y, Barr CL, Ren B (2016) A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep 17:2042–2059
Tanay A, Cavalli G (2013) Chromosomal domains: epigenetic contexts and functional implications of genomic compartmentalization. Current Opinion in Genetics & Development 23:197–203
The ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Valton AL, Dekker J (2016) TAD disruption as oncogenic driver. Curr Opin Genet Dev 36:34–40
Acknowledgements
Work supported by grants to MN from the NIH ID 1U54DK107977-01 and CINECA ISCRA ID HP10CYFPS5 and HP10CRTY8P. MN also acknowledges computer resources from INFN, CINECA, and Scope at the University of Naples.
Author Contributions
M.N. designed the study; S.B., A.M.C., C.A., M.N. developed the project; A.M.C., C.A., S.B., A.E. run the computer simulations and performed the analyses; A.M.C., C.A., S.B., M.N. wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Responsible editor: Nick Gilbert and Davide Marenduzzo.
Simona Bianco, Andrea M. Chiariello, and Carlo Annunziatella have equal contribution.
Rights and permissions
About this article

Cite this article
Bianco, S., Chiariello, A.M., Annunziatella, C. et al. Predicting chromatin architecture from models of polymer physics. Chromosome Res 25, 25–34 (2017). https://doi.org/10.1007/s10577-016-9545-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-016-9545-5