
Abstract
Cholesterol, a principal constituent of the cell membrane, plays a crucial role in the brain by regulating the synaptic transmission, neuronal signaling, as well as neurodegenerative diseases. Defects in the cholesterol trafficking are associated with enhanced generation of hyperphosphorylated Tau and Amyloid-β protein. Tau, a major microtubule-associated protein in the brain, is the key regulator of the mature neuron. Abnormally hyperphosphorylated Tau hampers the major functions related to microtubule assembly by promoting neurofibrillary tangles of paired helical filaments, twisted ribbons, and straight filaments. The observed pathological changes due to impaired cholesterol and Tau protein accumulation cause Alzheimer's disease. Thus, in order to regulate the pathogenesis of Alzheimer's disease, regulation of cholesterol metabolism, as well as Tau phosphorylation, is essential. The current review provides an overview of (1) cholesterol synthesis in the brain, neurons, astrocytes, and microglia; (2) the mechanism involved in modulating cholesterol concentration between the astrocytes and brain; (3) major mechanisms involved in the hyperphosphorylation of Tau and amyloid-β protein; and (4) microglial involvement in its regulation. Thus, the answering key questions will provide an in-depth information on microglia involvement in managing the pathogenesis of cholesterol-modulated hyperphosphorylated Tau protein.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cholesterol, a structurally important component of cellular membrane and myelin sheath, is the principal constituent of the brain. In comparison to the whole body, the brain contains 20% of the body’s total cholesterol (Björkhem et al. 2006). Glycerophospholipids, sphingolipids, and cholesterol are the three major brain lipid components (Korade and Kenworthy 2008). Neurons and glia (astrocytes, microglia, and oligodendrocytes) are helping to regulate the cholesterol concentration to achieve healthy brain development. The critical functions of the brain cholesterol include synapse, dendrite formation, and axonal guidance. Along with this, brain cholesterol also contributes in precursor activity, steroid hormones, and bile acid synthesis (de Chaves et al. 1997; Radhakrishnan et al. 2007; Fester et al. 2009; Wong et al. 2014). Bile acids are the amphipathic end products of cholesterol metabolism. It plays a significant role in cell signaling by binding to cell membrane and nucleus. Also, it has been reported to interact with receptors involved in neurotransmission (McMillin and DeMorrow 2016). The impairment of synaptic vesicle exocytosis, neuronal activity and neurotransmission, dendritic spine, and synapse degeneration due to cholesterol depletion in neurons causes Alzheimer’s, Parkinson's, Niemann–Pick C, and Huntington’s diseases (Block et al. 2010; Linetti et al. 2010; Di Paolo and Kim 2011; Wang et al. 2011). Alzheimer’s disease (AD), one of the multifactorial neurodegenerative diseases, is characterized by pathological features of cerebral atrophy, extracellular amyloid-β (Aβ) plaques, intracellular neurofibrillary tangling, neurovascular dysfunction, and neuronal cell death (Sato and Morishita 2014; Ramanathan et al. 2015). The population suffering from AD have a progressive decline in memory, thinking, language, and learning capacity (Duthey 2013). Nearly 8–10% of the population over the age of 65 suffer from AD, and in every 5 years, the number is getting doubled (Cummings 2004; Bertram and Tanzi 2005). Most of the patients suffering from AD are sporadic, whereas 5% or less are familial with an early-onset issue (Kang and Rivest 2012). The early-onset form of FAD is linked with mutations in Amyloid Precursor Protein (APP), presenilin-1 (PS-1), and presenilin-2 (PS-2) affecting APP processing which results in increased production of insoluble Aβ plaques. The overproduced Aβ plaques, in turn, cause pathogenic Tau hyperphosphorylation which leads to neurodegenerative diseases (Selkoe and Hardy 2016; Szaruga et al. 2017). Sporadic AD being the late-onset AD (LOAD) appears mainly due to genetic and environmental factors including aging that impair the brain’s ability to clear Aβ and Tau protein (Wildsmith et al. 2013; Pereira et al. 2019). Hence, based on the prevalence, AD is considered as one of the most significant health and socio-economic problems (Wong et al. 2014). Initially, full-length APP localized in the plasma membrane, as well as several organelles like mitochondria, endoplasmic reticulum (ER), and Golgi apparatus, was considered as the primary reason for the development of AD. Thus, anti-amyloid strategies have been the primary focus of AD drug development for the last 10 years. But recently, more and more evidence has demonstrated the crucial role of Tau abnormalities in AD neurodegeneration (Gong and Iqbal 2008; Busche et al. 2019). This review aims to explore the detailed molecular mechanisms of Taupathy in AD and its regulation via microglial uptake.
Tau Protein
Tau, a microtubule-associated protein (MAP) present in the central nervous system (CNS), is associated with several functions related to neuronal cells. Tau gene is located at chromosome 17q21, comprising 16 exons, among which 1, 4, 5, 7, 9, 11, 12, and 13 are constitutive, whereas 2, 3, and 10 undergo alternative splicing. The dysregulation of alternative splicing process especially in exon 10 contributes to neurodegeneration (Andreadis 2005; Liu and Gong 2008). Axonal transportation, neurotransmission and assembly, and stability of microtubules are some of the major functions of Tau protein (Iqbal et al. 2005a). Tau undergoes various posttranslational modifications like phosphorylation, glycosylation, and ubiquitinylation (Morris et al. 2015; Avila et al. 2016; Wang and Mandelkow 2016; Sonawane and Chinnathambi 2018). Hyperphosphorylation of Tau hampers its microtubule assembly activity as well as binding capability (Lindwall and Cole 1984; Alonso et al. 1994). Among several reasons, cholesterol metabolism in the brain is one of the major causes of Tau hyperphosphorylation (Rahman et al. 2005; Michikawa 2006; Maccioni et al. 2010). Tau is hypothesized to get released from its intracellular localization into extracellular space. The extracellular released Tau is reported to cause severe damage due to its interaction with neighboring cells, such as neurons or glia (Luo et al. 2015; Hu et al. 2016; Bolós et al. 2017b). Thus, regulating these hydrophilic soluble proteins (Tau) is of major concern. Microglia, the macrophages of the brain, are the resident immune cells constituting about 15% of cell population in CNS (Lawson et al. 1992). It was discovered between 1919 and 1921 by a Spanish neuroscientist, Pío del Río Hortega, and was hypothesized to undergo morphological changes, proliferate, and migrate with respect to change in microenvironment (Pérez-Cerdá et al. 2015; Perea et al. 2018). Microglia are reported to play a major role in interacting, internalizing, and regulating the pathological Tau (Luo et al. 2015; Perea et al. 2018; Hopp et al. 2018). Thus, the current review article is focused on emphasizing the mechanism of Tau pathology due to cholesterol metabolism and their regulation via microglia in managing the AD condition.
Cholesterol Biosynthesis in Neurons and Astrocytes
Cholesterol, an indispensable component of biological membranes, acts as precursor for various signaling molecules. Except brain, its provision and disposal in all other organs of the mammalian body rely upon dietary intake, on de novo synthesis in every organ, and lipoprotein-mediated transport via the blood circulation (Pfrieger and Ungerer 2011). But, the cells in the brain are cut off from this process due to blood–brain barrier (BBB) and thus they have adopted their own specific way to handle cholesterol turnover. Therefore, the necessary cholesterol in the brain is synthesized de novo within CNS (Gamba et al. 2015). Brain cholesterol, considered as a distinct pool from body cholesterol, is majorly composed of non-esterified cholesterol with small amounts of desmosterol and cholesteryl ester (Dietschy and Turley 2001; Björkhem and Meaney 2004). The amount of cholesterol in the brain is high (15–30 mg/g tissue) compared to other tissues of the body (2–3 mg/g tissue) (Dietschy 2009). The importance of brain cholesterol has been described in early 1834’s as “un element principal” of the CNS (Couerbe 1984). Brain cholesterol is found in two major forms, myelin sheath and plasma membrane of neurons and glial cells. The smaller portion of cholesterol constitutes to produce plasma membrane of neurons (10%) and glial cells (20%) and have relatively fast turnover rates of 5–10 months, and 8 mg/g half-life. However, myelin sheath, a protective outer membrane of the neurons, constitutes the major portion of cholesterol (70–80%) that is composed of oligodendrocytes have slower turnover number (half-life ~ 5 years, 40 mg/g). These are reported to maintain the morphology and synaptic transmission (Dietschy and Turley 2004). Once the myelin sheath encircles the neurons, the majority of brain cholesterol accumulates between perinatal and adolescence period (Orth and Bellosta 2012). After myelination, very low turnover and loss is observed in the adult brain (Morell and Jurevics 1996). Brain cholesterol is more stable than the peripheral with the turnover rates of 1% (Andersson et al. 1990). The half-life of adult brain cholesterol is between 6 months to 5 years, whereas the plasma cholesterol is only for few days (Dietschy and Turley 2004; Björkhem et al. 2006).
The de novo biosynthesis of cholesterol in the brain is a complex and intense process. Acetyl CoA, a primary precursor molecule, is converted to 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) by 3-hydroxyl-3-methylglutaryl-CoA synthase (HMG-CoA synthase). HMG-CoA is further reduced to mevalonate by HMG-CoA reductase. The same is considered as the rate-limiting and irreversible step in cholesterol synthesis. After a series of nearly 20 enzymatic reactions, cholesterol formation is observed (Zhang and Liu 2015). The other important metabolites along with cholesterol include mevalonate, farnesyl pyrophosphate, squalene, and lanosterol (Bae et al. 1999). The necessary enzymes involved in de novo synthesis occur primarily in the ER, which are further transferred to the plasma membrane (DeGrella and Simoni 1982; Gaylor 2002). The redistribution of cholesterol in the subcellular compartments is accomplished by two ATP-driven processes, i.e., vesicles-mediated inter-organelle transport and protein-mediated monomeric transport/non-vesicular transport (Kaplan and Simoni 1985; Heino et al. 2000). The energy-dependent vesicular transport contributes to cholesterol flux between ER, plasma membrane, and endocytic vesicles (Litvinov et al. 2018). However, the non-vesicular transport along with carrier proteins or membrane contact between mitochondria and lipid droplets helps in the circulation of cholesterol in the outer surface (Kallen et al. 1998). Since cholesterol is water-insoluble, the amount of unbound cholesterol in the cytosol is minimal. Thus, most of the cholesterol exists as protein-bound in the cytosol as apolipoprotein E (APOE) (Zhang and Liu 2015). There are two cholesterologenic pathways in the brain, sterols in the neurons are synthesized via the Kandutsch–Russell pathway, and astrocytes contain precursors from the Bloch pathway (Leoni and Caccia 2015). The post-squalene ancestor, and lanosterol obtained during the cholesterol synthesis in the brain, acts as a precursor molecule for the cholesterol synthesis in astrocytes and neurons (Zhang and Liu 2015).
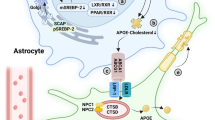
The Kandutsch–Russell pathway employed in the neurons converts the precursor molecule, lanosterol, into 24, 25-dihydroxylanosterol. Further reduction in 24, 25-dihydroxylanosterol is reported to generate cholesterol. In case of Bloch pathway, astrocytes convert the post-squalene component, lanosterol, into 32-hydroxylanosterol. The later consecutive reduction steps with various enzymes end up in the generation of cholesterol from desmosterol. Astrocytes meet neuronal cholesterol demand by secreting APOE–cholesterol complexes, which are then transported to the neurons for their development and function (Swathi Parasuraman 2013). Figure 1 delineates the de novo synthesis of cholesterol in brain and its adaptive pathways to manage its turnover via neurons and astrocytes.

Cholesterol synthesis in the brain, neuron, and astrocytes. Cholesterol in the brain is primarily synthesized from the precursor molecule acetyl-CoA by a series of enzymatic steps. Initially, acetyl-CoA gets transformed into 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) via a catalyzed reaction by HMG-CoA-synthetase and then by HMG-CoA reductase to mevalonate. 3-hydroxy 3-methyl-glutaryl-CoA reductase (HMG-CoA reductase) acts as rate-limiting step for mevalonate pathway. This is followed by a series of enzymatic reactions in converting mevalonate into squalene, lanosterol, and other products. In the final step, 7-dehydrocholesterol is converted to cholesterol by the enzyme 7-dehydrocholesterol reductase. Finally, the hydroxylated derivative of cholesterol, 24-hydroxycholesterol, produced by CYP46, acts as the only molecule capable of crossing the BBB. The maximum production of brain cholesterol is observed during the perinatal period, mainly for myelination. The post-lanosterol steps of cholesterol synthesis are divided into Bloch and Kandutsch–Russell pathways. Cholesterol in neurons is primarily biosynthesized through Kandutsch–Russell pathway by consecutive reaction steps involving 24, 25-dihydroxylanosterol, zymosterol, lanosterol, 7-dehydrocholesterol. In case of astrocytes, the Bloch pathway utilizes the precursor molecule lanosterol from the brain, hence converting to 32-hydroxylanosterol, zymosterol, cholestadienol, 7-dehydrodesmosterol, desmosterol, and cholesterol. Further, the excess cholesterol synthesized from astrocytes is supplied to the brain to maintain the cholesterol turnover
Cholesterol Trafficking Between Astrocytes and Neurons
Neuronal cells regulate the cholesterol content by balancing the biosynthesis, import, and excretion via the feedback mechanism. The biosynthesis of cholesterol occurs in the ER and further transferred to the Golgi apparatus and plasma membrane (Rogers et al. 2015; Xu et al. 2018). The membrane-bound transcription factors such as sterol regulatory element-binding proteins (SREBPs), SREBP1 and SREBP2, act as a tool in helping the cells to balance the cholesterol content by regulating the transcription of genes encoding enzymes related to cholesterol, fatty acid biosynthesis, as well as lipoprotein receptors (Eberlé et al. 2004). The maximum amount of cholesterol within CNS is associated with the cell membrane, thus leaving behind a minimal amount in the intercellular space and the cerebrospinal fluid. APOE that is highly expressed in the intercellular space of the CNS helps in associating the unbound cholesterol, thus regulating its metabolism, as well as genetic risk factors related to sporadic AD (Ashford 2004). Also, the astrocytes or the glial cells present in the brain are reported to help cholesterol recycling in the peripheral nervous system. The lipoprotein particles secreted by astrocytes are also composed of APOE, APOA1, and lipids (Orth and Bellosta 2012). A 32 kDa APOE gene residing on chromosome 19 contains three major form of alleles, i.e., ε2 (5–10%), ε3 (65–70%), and ε4 (15–20%) (Orth et al. 1999; Bu 2009; Holtzman et al. 2012). Other than astrocytes, oligodendrocytes, microglia, and ependymal layer cells are also reported to be the major sources of APOE (Mahley et al. 2006). The APOE-containing lipoproteins secreted by glial cells, lipids, and other macromolecules act as ligands for numerous lipoprotein receptors of low-density lipoprotein (LDL), very low-density lipoprotein receptor (VLDLR), and LDL receptor-related protein 1 (LRP1) family (Liu et al. 2007b; Pottier et al. 2012). Since APOE is the major exporter of extracellular cholesterol and other lipids, dynamic exchange of APOE has been reported among cells (Lahiri 2004). APOE2 defectively binding to LDL receptor is responsible for type III hyperlipidemia. APOE3 and APOE4 are also reported to be highly associated with LDL receptor (Mahley 2016). Chances of developing LOAD are high in individuals with one ε4 allele than with those without the ε4 allele. And people having ε2 allele are reported to be at lower risk in developing LOAD compared to ε3 allele (Corder et al. 1993). The in vivo study conducted on mice expressing P301L mutant human Tau with no apoE and P301L mice with wild-type ApoE fed with cholesterol-enriched diet or control diet for 15 weeks showed enhanced ongoing Tau pathology only in wild-type mice, thus contributing to AD pathology (Glöckner et al. 2011). In addition to APOE, genome-wide association studies (GWAS) have identified novel risk genes of AD. Triggering receptor expressed on myeloid cells 2 (TREM2) in the brain is expressed primarily by microglia. It is reported to promote microglial survival, proliferation, and phagocytosis of excess cholesterol (Wolfe et al. 2019). The most common TREM2 mutant, R47H (arginine to histidine at position 47), impairs ligand binding and increases the risk of developing AD by approximately fourfold (Guerreiro et al. 2013a; Jonsson et al. 2013). Other than this, some of the identified genes are reported to be associated with lipid metabolism. Also, diabetic patients affected from glucose-energy metabolism are reported to be significantly at higher risk of developing AD (Maher and Schubert 2009; Matsuzaki et al. 2010; Hollingworth et al. 2011). Other signaling pathways like ATP-binding cassette transporter (ABCA1), also known as a cholesterol efflux regulatory protein (CERP) with the MAP-kinase system, are also active in microglia and neurons (Witzlack et al. 2007). ATP-binding cassette (ABC) transporters, namely ABCA1, ABCG1, and ABCG4, expressed in neurons and astrocytes mediate sterol efflux at the plasma membrane (Tarr and Edwards 2008; Kim et al. 2008). Among them, ABCA and ABCG play a major role in regulating the lipid homeostasis (Puglielli et al. 2003). Expression of ABCA1 has been reported in both neurons and glial cells, whereas a higher level is in neurons, thus contributing to cholesterol efflux via APOA1.
The excess cholesterol is released from APOA1-containing lipoproteins to LRP1/LDLR receptor located in neurons (Gosselet et al. 2009). In comparison to LRP1, LDLR is highly expressed in glial cells than neurons (Rebeck et al. 1993), whereas the highly expressed LRP1 in neurons contributes to the transportation of APOE-associated cholesterol from astrocytes (Li et al. 2001). Downregulation of ABCA1 and LRP1/LDLR expression reduces the cholesterol efflux. However, increased level of the receptors maintains the excess cholesterol either by esterification or hydroxylation process catalyzed by acyl-coenzyme A: cholesterol acyltransferase-1 (ACAT1/SOAT1) present primarily in the ER (Wüstner et al. 2005; Minagawa et al. 2009). The externally uptaken excess cholesterol enters subcellular endosomal membrane compartments like Niemann–Pick type C1 (NPC1) & NPC2 (Vanier 2015). The transmembrane protein NPC1 is associated with the sterol-sensitive domain, whereas NPC2 being an intraluminal component binds to cholesterol (Carstea et al. 1997; Soccio and Breslow 2004). The dysfunction of either the protein causes accumulation of unesterified cholesterol, thus causing pathological changes in neurons and glial cells (Reid et al. 2004). Also, the presence of excess unesterified cholesterol contributes to the excessive movement of cholesterol between the plasma membrane and ER, as well as genetically impairs the ACAT1/SOAT1 gene (Wollmer et al. 2003; Hutter-Paier et al. 2004; Karten et al. 2006).
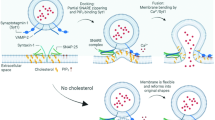
The surplus amount of esterified cholesterol in neurons is stored as lipid droplets or excreted out from the brain as 24-hydroxycholesterol (24-OHC) via hydroxylation process at the rate of 6–12 mg/day (0.02–0.04% of the total cholesterol turnover) (Dietschy 2009; Bryleva et al. 2010). The cholesterol 24-hydroxylase enzyme catalyzing the above reaction is located in somata, dendrites of neurons, as well as in the glial cells of the brain. Thus, the major cholesterol turnover is observed in the brain than astrocytes (Ramirez et al. 2008). The hydroxylated product, 24-OHC, is reported to up-regulate the expression of ABCA1 via activated liver X receptor (LXR) in astrocytes and neurons (Gabbi et al. 2014; Korach-André and Gustafsson 2015). LXR acts as a cholesterol sensor by protecting the cell from excess internal cholesterol by suppressing cholesterol biosynthesis via cholesterol efflux by feedback process (Wang et al. 2008; Kang and Rivest 2012), stimulates reverse cholesterol transport (RCT) from peripheral tissue, activates the conversion of cholesterol, and decreases intestinal cholesterol absorption (Baranowski 2008). Hyperphosphorylation of Tau protein due to impaired cholesterol accumulation caused by overexpression/mutation in the functioning of cell-surface receptors such as ATP-binding cassette transporters, LRP1/LDLR, and endosomal membrane compartment-NPC1/2 leads to neurodegenerative disorders (Akram et al. 2010; Rodrıguez-Rodrıguez et al. 2010; Liu et al. 2010; van der Kant et al. 2019). Thus, cholesterol homeostasis between astrocytes and neurons plays a crucial role in modulating the pathogenesis in brain (Fig. 2).

Cholesterol homeostasis in astrocytes and neurons, correlation with Tau protein hyperphosphorylation. Although neurons are capable of synthesizing cholesterol, it has been suggested that neurons rely on astrocytes for cholesterol delivery in the adult state. The major input of cholesterol into the brain comes from in situ synthesis in the ER of astrocytes. Lipoprotein particles, including apolipoprotein E (APOE) assembled in the ER and the newly synthesized cholesterol, are transported from the ER to extracellular space by non-vesicular mechanism via ATP-binding cassette transporter (ABCA1). The cholesterol-rich APOE particles interact with neuronal receptors LRP1/LDLR and undergo internalization by receptor-mediated endocytosis or routed to late endosomes/lysosomes internalization into neurons. Endosome/lysosome in assistance of NPC1/C2 leads to the generation of free cholesterol. The obtained free cholesterol is converted to 24-ortho-hydroxy cholesterol (24-OHC), which can pass through the BBB. Binding of 24-OHC to the cytoplasmic liver X receptors triggers expression of ABCA1, ABCG1, and APOE genes involved in cholesterol trafficking from astrocytes to neurons. The excess cholesterol in the neurons is stored as lipid droplets in the neurons. Although the BBB is not permeable to plasma cholesterol, BBB endothelial cells make possible cholesterol flux across via ABC transporters and LRP1 receptors. Non-functioning or mutation in ATP-binding cassette transporter, LRP1/LDLR, and Niemann–Pick C1/C2 genes leads to the excess cholesterol accumulation, in turn causing Tau hyperphosphorylation
Tauopathies
The normal function of mature neurons is accomplished by some of the major MAPs like Tau, MAP1 (A/B), and MAP2. The primary role of these three MAPs is towards the promotion of assembly and stability of microtubules in the neurons. In spite of its importance, neurons can compensate for the loss of function of one MAP with the other two MAPs. According to Teng et al., healthy development in adult life was observed in both tau and MAP2 knockout transgenic mice. However, double knockout of Tau and MAP2, as well as MAP2 and MAP1B in transgenic mice, shows defects in axonal elongation and neuronal migration (Takei et al. 2000; Teng et al. 2001). Also, chemical or genetic activation of microglia in the Tau mouse model of Tauopathy is reported to significantly contribute Tau pathology (Bhaskar et al. 2010). The major biological activity of Tau protein is regulated by the degree of phosphorylation. The optimal activity of Tau protein is achieved by 2–3 mol phosphate/mol of the protein (Köpke et al. 1993). The abnormally hyperphosphorylated Tau protein (~ three to fourfold higher) is reported to be accumulated as interneuronal tangles of paired helical filaments (PHFs), twisted ribbons, or straight filaments in AD (Wegmann et al. 2010), thus leading to neurofibrillary degeneration and dementia due to reduced microtubule assembly and binding capability (Brier et al. 2016). Phosphorylated Tau protein in AD is a substrate for several protein kinases (Singh et al. 1994; Johnson and Hartigan 1999). Exogenous cholesterol, glycogen synthase kinase-3 (GSK-3), cyclin-dependent protein kinase-5 (cdk5), protein kinase A (PKA), calcium and calmodulin-dependent protein kinase- II (CaMKII), mitogen-activated protein kinase ERK 1/2, and stress-activated protein kinases are the major contributors for abnormal Tau hyperphosphorylation (Pei et al. 2003). Tau phosphorylation at different sites has shown the disparate impact on its biological function and pathogenesis. According to an in vitro study, Tau phosphorylation at Ser262, Thr231, and Ser235 inhibits its binding to microtubules by ~ 35%, ~ 25%, and ~ 10%, respectively (Sengupta et al. 1998). However, the in vitro kinetic studies related to the binding of hyperphosphorylated and normal Tau suggest that phosphorylation at sites, Ser199/Ser202/Thr205, Thr212, Thr231/Ser235, Ser262/Ser356, and Ser422 sequesters normal MAPs from microtubules (Alonso et al. 2004). Further self-aggregation of Tau into filaments is promoted by phosphorylation at Thr231, Ser396, and Ser422 sites. Also, mutation of Tau at Ser396 and Ser404 into Glu to mimic phosphoserine converts it to be more fibrillogenic (Abraha et al. 2000). According to the reports, increased propensity of Tau protein aggregation is observed due to the mutation at Ser422 to Glu (Haase et al. 2004).
The major contributors of Tau hyperphosphorylation are exogenous cholesterol and the increased serum cholesterol. The NPC2 characterized by the accumulation of exogenous cholesterol in lysosomes due to defective sorting/trafficking mechanism is reported to be associated with Tau hyperphosphorylation (Fan et al. 2001). Also, defective assembly of cholesterol and phospholipids within APOE due to mutated ABCA1 leads to Tau hyperphosphorylation (Akram et al. 2010). The two major lipoprotein receptors LRP1 and LRP2 bind and internalize extracellular ligands for degradation in lysosomes. The lipoprotein receptors also mediate the cholesterol uptake into the neuron. Malfunction of the receptors impairs intracellular signaling as well as storage and/or release of cholesterol in neurons. Thus, the impaired cholesterol metabolism due to LRP1 inactivation also contributes to Tau hyperphosphorylation (Spuch et al. 2012). Proline-directed protein kinases are also reported for its contribution in abnormal hyperphosphorylation of Tau (Liu et al. 2007a). As reported by Iqbal et al., GSK-3β, cdk5, and ERK ½ are the three major proline-directed protein kinases associated with Tau phosphorylation at specific sites in AD (Iqbal et al. 2005b). Also, as reported in a triple transgenic mouse model of AD, inflammation via toll-like receptor-4 (TLR4) ligand contributes to hyperphosphorylation of Tau (Kitazawa et al. 2005). The attenuation or exacerbation of Tau pathology has been reported through blockage or activation of interleukin-1 signal (Kitazawa et al. 2011). In addition to damaging neurons through phagocytosis of synapses and worsening of Tau pathology, microglia can also react to protein aggregates and inflammatory mediators. Other contributors to Tau hyperphosphorylation include TNF-α, IL-1β, inflammatory cytokines produced from activated TLRs and NLRP3 inflammasome (Heneka et al. 2013, 2015).
CX3CR1, a CX3C chemokine receptor-1, also known as fractalkine receptor or G-protein-coupled receptor 13 plays a major role in the microglial migration and phagocytosis in the CNS (Paolicelli et al. 2011). The deficiency of CX3CR1 is reported to result in the acceleration of Tau pathology in a hTau mouse model (human Tau-containing transgenic mice) (Bhaskar et al. 2010; Cho et al. 2011). CX3CL1 produced extensively in the neurons binds to the receptor CX3CR1, which is expressed on the surface of microglia (Kim et al. 2011; Zhan et al. 2014). Tau protein has also been reported binding to CX3CR1, thus increasing its internalization by microglia. Hence, increased competition has been observed between Tau and CX3CL1 in binding to the receptor (Bolós et al. 2017a). Deletion of microglial protein CX3CR1 in transgenic Tau model has shown genetic enhancement in the microglial activation and progression of Tau pathology. Thus, the reduced microglial phagocytic activity induces Tau hyperphosphorylation (Bhaskar et al. 2010; Maphis et al. 2015; Perea et al. 2018). The abnormally hyperphosphorylated Tau has been reported to develop resistance to proteolysis by the calcium-activated neutral proteases, thus leading to tauopathy in AD patients due to its lower turnover number (Khatoon et al. 1992; Wang et al. 1996; Poppek et al. 2006). Similarly, according to Maphis et al., a significant accelerated onset and progression of tau pathology, cognitive dysfunction, and neurodegeneration were observed in hTau CX3CR1−/− mice due to genetic enhancement of microglia-specific neuroinflammation. The microglial activation contributed to the spread of tau pathology through anatomically connected neurons of the hippocampus. Also, the adoptive transfer of purified microglia from hTau CX3CR1−/− mice to non-transgenic mice showed Tau hyperphosphorylation, thus indicating the role of microglia and its receptor, CX3CR1, in the dissemination of Tau pathology in the brain (Maphis et al. 2015).
As previously described, TREM2 functioning similar to APOE ε4 is reported to regulate inflammatory responses of microglia and phagocytosis of cellular debris (Li and Zhang 2018). Mutation in the TREM2 cell-surface receptor at R47H position impairs TREM2-mediated microglial activation of phagocyting hyperphosphorylated Tau protein (Guerreiro et al. 2013b; Jonsson et al. 2013; Jay et al. 2017; Ulrich et al. 2017). Also, TREM2-deficient microglia shows reduced uptake of Tau–cholesterol complex, thus leading to enhanced accumulation and risk of AD (Yeh et al. 2016). The impact of TREM2 deficiency investigated in the brain of young and aged mice using RNA-sequencing reported the pathogenesis of AD due to disrupted immune response (Carbajosa et al. 2018).
Some of the other factors influencing the Tau pathology include insulin resistance (Starks et al. 2015). Glucose uptake and metabolism impairment have been reported in AD brain (Hoyer 2004). The impaired glucose uptake/metabolism causes deregulation of Tau phosphatases and decreased Tau O-GlcNAcylation, thus facilitating abnormal hyperphosphorylation of Tau protein in many diabetic animal models (Gong et al. 2006; Jolivalt et al. 2008; Ke et al. 2009; Qu et al. 2011).
GSK-3, a proline-directed serine/threonine kinase, is reported to play a pivotal role in the pathogenesis of both sporadic and familial forms of AD (Hooper et al. 2008). The activity of GSK-3 is reported to be regulated by insulin and Wnt signaling pathways. Insulin signaling leads to the activation of PI3-kinase, which in turn activates Akt protein. According to the reported studies, the activated Akt phosphorylates free cytoplasmic GSK3β and GSK3α at serine (Ser) residues 9 and 21, respectively (Saltiel and Kahn 2001; Lizcano and Alessi 2002). Both GSK-3 α and β analogs play a major role in the hyperphosphorylation of Tau protein, memory impairment, increased production of Aβ, and inflammatory response (Hooper et al. 2008). Attenuation of phosphoinositide 3-kinase/Akt (PI3k/Akt) signal was observed in the brain due to diet-induced insulin resistance. The increased GSK-3α activity observed by attenuating GSK-3β phosphorylation from insulin resistance resulted in increased Aβ production (Ho et al. 2004). Insulin resistance together with oxidative stress causes inhibition of Wnt signaling, which in turn is reported to activate GSK-3β (Manolopoulos et al. 2010). The inhibited Wnt thus promotes the pathogenesis of Aβ and Tau protein hyperphosphorylation (Phiel et al. 2003; Rankin et al. 2007). In addition to its role in increasing Aβ production, insulin resistance is reported to reduce Aβ clearance by decreasing the expression and activities of insulin-degrading enzyme (IDE) (Ho et al. 2004). Thus, by regulating the activity of IDE, Aβ concentration can be reduced. The overview of the influencing parameters involved in the hyperphosphorylation of Tau protein, as well as Aβ accumulation, is summarized in Fig. 3.

Parameters involved in the hyperphosphorylation of Tau protein in microglia. Microtubule-associated protein, Tau, involved in the microtubule dynamics, treadmilling, cargo transport, and axonal out growth, becomes aggregated into senile plaques in the brain under diseased conditions. Tau hyperphosphorylation is associated with impairment of cholesterol metabolism via non-functioning and mutations of ATP-binding cassette transporter, LRP1 receptor, Niemann–Pick C1/C2 genes, surface receptor CX3CR1, triggering receptor expressed on myeloid cells 2 (TREM2 involved in regulating microglia and phagocytosis), inflammatory cytokine, NF-κB activation, as well as glycogen synthase kinase-3 (GSK-3) activation via insulin signaling impairment
Molecular Mechanisms of Microglial Uptake of Cholesterol, Tau, and Amyloid-β protein
Microglia are involved in the normal development, function, and repair of CNS. During injury or other pathological conditions, microglia migrate to lesion sites and initiate the immune response and resolve the particular insults in both human and transgenic murine models of AD (Nayak et al. 2014; Colonna and Butovsky 2017). As per previous reports, neurodegenerative diseases such as AD and Parkinson’s are associated with abnormal intracellular accumulation of Tau protein (Ballatore et al. 2007; Goedert and Spillantini 2011; Arendt et al. 2016). The normal function of Tau, i.e., promoting and stabilizing microtubules is disrupted to form aggregates and neurofibrillary tangles due to hyperphosphorylation (Iqbal et al. 2009; Medeiros et al. 2011). The mice with mutant human Tau are reported to be associated with high levels of cellular cholesterol, thus leading to Tau hyperphosphorylation (Maccioni et al. 2010; Glöckner and Ohm 2014). The hyperphosphorylated accumulated Tau protein gets released outside the cells into the extracellular space, thus interacting with other cells and causing the cell-to-cell transfer (Simón et al. 2013; Medina and Avila 2014; Bolós et al. 2016). Microglia, the brain macrophages, play a major role in regulating the excess cholesterol and hyperphosphorylated Tau by activation/phagocytosis function (Hansen et al. 2017). The living neurons with Tau inclusions from P301S-Tau mice were reported to be phagocytized by BV2 cells or primary microglia cells (Brelstaff et al. 2018). Recently, direct internalization of Tau protein by microglia was observed in an in vitro study. The phagocytosis of newly synthesized full-length Tau oligomer hTau40WT was evidenced with A11+ Iba1high microglial activation (Das et al. 2020). Similarly, the microglial engulfment of insoluble Tau protein obtained from homogenate of post-mortem AD brain tissue was observed in both in vitro and in vivo studies (Bolós et al. 2016). In the last decade, human genetic studies, especially GWASs using single-nucleotide polymorphisms (SNPs), have identified over 20 genetic loci that are robustly associated with AD risk (Lambert et al. 2013; Karch et al. 2014). A cell-surface transmembrane receptor protein TREM2 of the immunoglobulin superfamily is reported to be highly expressed both by the microglia in the brain, as well as by certain myeloid cells in the periphery (Guerreiro et al. 2013b; Jonsson et al. 2013). It acts as a cell-surface receptor via its interaction with the activating adaptor protein DAP12 (encoded by TYROBP gene). Various moieties activating the signaling pathways reported to be associated with TREM2 include lipopolysaccharides, phospholipids, HDL, LDL, APOE, APOJ, apoptotic neurons, Tau protein, and Aβ protein (Wolfe et al. 2019). Also, the stimulated TREM2 initiates various signal transduction pathways related to microglial chemotaxis, phagocytosis, survival, and proliferation (Mazaheri et al. 2017; Zheng et al. 2017). The cholesterol and hyperphosphorylated Tau protein are more efficiently taken up by microglia only when complexed with lipoproteins such as LDL, APOE, and CLU/apoJ (Terwel et al. 2011; Yeh et al. 2016). Besides TREM2, many other genes such as CD33, INPP5D, MS4A6A, and PLCG2 are also expressed in the microglia.
TREM2 is involved in the recruitment of tyrosine kinase SYK, which in turn phosphorylates immunoreceptor tyrosine-based activation motif (ITAM) of DAP12. The phosphorylated ITAM motif thereby activates downstream effectors such as PI3K and Ca2+ signaling. Potential-dependent Ca2+ channels and synaptic vesicles fuse with the presynaptic membrane to exocytosis excess cholesterol and hyperphosphorylated Tau protein (Zefirov and Petrov 2010). Another gene INPP5D, which encodes for lipid phosphatase SHIP1, is expressed in the microglia and this enzyme interacts with ITAM of DAP12 and dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) to phosphatidylinositol (3,4)-bisphosphate (PIP2) at the plasma membrane, thus altering the outcomes of PI3K activation (Peng et al. 2010; Boucrot et al. 2015). PIP3 acts as a secondary messenger in phosphorylating and activating AKT (serine/threonine protein kinase B), which in turn helps in regulating cell apoptosis, promoting protein synthesis as well as inhibiting FOXO protein (Manna and Jain 2015). Some of the inhibitory receptors, like CD33, a member of sialic acid-binding immunoglobulin-type lectins (SIGLEC) family receptors, are reported to inhibit the activity of ITAM and SYK (Malik et al. 2013; Raj et al. 2014), whereas the protective SNP alters CD33 mRNA splicing, thus restoring the TREM2-ligand binding, ITAM signaling, and phagocytosis activity (Griciuc et al. 2013; Bradshaw et al. 2013).
The Bridging Integrator 1 (BIN1) located on 2q14.3 chromosome is reported to be key epigenetic regulator within the cell. BIN1-amphiphysin2 plays a key role in mediating cell processes such as from endocytosis to membrane recycling and cell cycle progression to apoptosis (Prokic et al. 2014). It is regarded as the second most important genetic risk factor for LOAD after APOE ε4 (Naj et al. 2011). Low expression of BIN1-amphiphysin2 observed in the diseased brains is reported to promote Tau pathology, whereas overexpression inhibits the above process by promoting endocytosis process (Tan et al. 2013; Calafate et al. 2016). It is reported to modulate Tau pathology in addition to Aβ protein by co-localizing and interacting with RIN3 (a guanine nucleotide exchange factor for Rab5 and Rab31) (Kajiho et al. 2011; Chapuis et al. 2013; Holler et al. 2014; Zhou et al. 2014). In humans, totally 1–7 isoforms are reported to specifically get expressed in brain, isoform 8 in skeletal muscles, and isoforms 9 and 10 are ubiquitous (Ellis et al. 2012; Prokic et al. 2014). The BAR domain expressed with many proteins involved in membrane dynamics in the cells acts as the key modulators for all BIN1 isoforms to bind dynamin through the SH3 domain. But, only the neuronal isoforms of BIN1 containing a CLAP domain mediate the interaction of clathrin and AP2, and help clathrin-mediated endocytosis (CME) of Tau protein in the brain (Ramjaun and McPherson 1998; Slepnev et al. 2000). An adapter protein CD2AP, discovered as a ligand protein, interacts with T-cell-adhesion protein-CD2. It helps in membrane trafficking during endocytosis and cytokinesis (Wolf and Stahl 2003; Ma et al. 2010). CD2AP present between membrane proteins and actin cytoskeleton interacts with SHIP1 and RIN3, and regulates the microglial endocytosis of Tau protein (Bao et al. 2012; Rouka et al. 2015). The loss of CD2AP function has been reported to enhance Aβ metabolism, Tau-induced neurotoxicity, abnormal neurite structure modulation, and reduced BBB integrity (Shulman et al. 2014; Qing-Qing et al. 2018) (Fig. 4a).

Molecular mechanisms of microglial uptake of cholesterol, Tau, and amyloid-β protein. a Lipoproteins containing APOE or apoJ carrying cholesterol in association with Tau protein bind to TREM2. The ligand-activated TREM2 interacts with immune receptor tyrosine-based activation motifs (ITAMs), which leads to recruitment of spleen tyrosine kinase. The ITAM/SYK mediates the activation of phosphoinositide 3-kinase-AKT pathway and Ca2+ signaling. Through Ca2+-dependent channels, synaptic vesicles fuse with presynaptic membrane and thus exocytosis excess cholesterol and hyperphosphorylated Tau. SH-2-containing inositol 5′ polyphosphatase 1 (SHIP1) expressed on microglia interacts with ITAM and modulates CD2AP, RIN3, and BIN1 adaptor protein in regulating microglial endocytosis of Tau protein. b Regulation of Aβ protein occurs via binding to clathrin-dependent receptors, including CD33, SORL1, LRP1, and LRP1/HSPG-mediated uptake. The internalized Aβ traffics to lysosomes for subsequent degradation. LRP1 also controls the cytoskeleton by modifying PI3K regulation of RhoA and Rac1 in causing Aβ cellular uptake and degradation by macropinocytosis or phagocytosis process
Similarly, the extracellular peptide Aβ observed due to mutations within the genes encoding for PS-1 and PS-2 is cleared from the peripheral circulation via chaperone-mediated transport across the BBB (Shibata et al. 2000). Remarkably, wide varieties of genes are associated with Aβ processing or trafficking as well as myeloid cell-mediated Aβ clearance (Malik et al. 2015). LOAD risk genes related to increased complement activation and inflammation (APOE, INDPP5D, CR1, TREM2, MS4A), complement activation (CLU, CR1), the human leucocyte antigen (HLA) gene complex (HLA-DRB1, HLADRB5), myeloid cell-mediated Aβ proteolysis (ACE, CD2AP), and phagocytosis (APOE, BIN1, INPP5D, CR1, ABCA7, TREM2) are some of the important functions of the genes associated with Aβ protein (Karch and Goate 2015; Malik et al. 2015). The fundamental mechanism of Aβ clearance is either by removal to peripheral blood and lymphatic system or degradation within CNS tissues. Aβ reaches peripheral circulation via various mechanisms such as chaperone-mediated transport across the BBB (Shibata et al. 2000), perivascular drainage (Weller et al. 2008), or through the lymphatic system (Iliff et al. 2012; Iliff and Nedergaard 2013). The overexpressed small GTPases Rab5 and Rab7 promote the transfer of Aβ into lysosomes by regulating vesicle fusion (Li et al. 2012). The activated immune cells, mainly brain-resident microglia and infiltrating blood-borne monocyte-derived macrophages, play a crucial role in the physiological clearance of Aβ (Simard et al. 2006; Koronyo-Hamaoui et al. 2009). Transport across BBB requires special molecular chaperons belonging to LDLR family, such as LRP1 and ABC transporters (Tarasoff-Conway et al. 2015). LRP1 is the major clathrin-dependent endocytic receptor, located on the abluminal surface of brain endothelial cells. They stimulate the endocytosis of Aβ protein either by binding to APOE–Aβ complex or Aβ alone (Deane et al. 2004; Spuch et al. 2012). Once Aβ gets contained within the endothelial cells, the luminal transport protein ABCB1 facilitates the removal of Aβ into the vascular lumen via lysosomal degradation (Elali and Rivest 2013; Kanekiyo et al. 2013). Pharmacological inhibition of dynamin-mediated endocytosis leads to the prevention of transneuronal transmission of Aβ (Song et al. 2014). Receptor-associated protein (RAP), a chaperone and antagonist of LRP1, is also reported to interact with Aβ and facilitate its cellular uptake through heparin sulfate proteoglycan (HSPG) (Kanekiyo and Bu 2014). HSPG mediates the entry of various molecules including exosomes, cell-penetrating peptides, polycation-nucleic acid complexes, viruses, lipoproteins, growth factors, and morphogens into cells (Christianson and Belting 2014). It facilitates the initial binding of Aβ on the cell surface and further LRP1 mediates the endocytosis process by forming LRP1–HSPG complex (Wilsie and Orlando 2003; Kanekiyo et al. 2011). LRP1 also regulates Rac1 and RhoA activities in Schwann cell, which influences cell migration and adhesion (Mantuano et al. 2010). According to Yu et al., Aβ42 oligomers get internalized through a dynamin-dependent and RhoA-mediated endocytic pathway in neuronal cells. But no reports are available to describe the exact mechanism involved by LRP1 in activating RAC1 and RhoA (Yu et al. 2010). According to literature reports, LRP1 controls the cytoskeleton architecture by modifying PI3K/extracellular signal-regulated kinase (ERK) and/or focal adhesion kinase (FAK)/paxillin pathways. Thus, it has been predicted that the process mentioned above might contribute in regulating Rac1 and RhoA (Dedieu and Langlois 2008). The phosphatidylinositol binding clathrin assembly protein functions as an adaptor protein for the transcytosis of Aβ–LRP1 complex across BBB (Carrasquillo et al. 2010; Tian et al. 2013). Sortilin-related receptor 1 (SORL1) (also known as SorLA and LR11) are reported to be involved in the intracellular transport and processing of APP, thus resulting in decreased production of Aβ peptide (Andersen et al. 2005; Fjorback et al. 2012). Disruption of SORL1 influences the Aβ pathway and Tau-related cellular processes by mediating their endocytic pathways (Aβ and Tau) (Offe et al. 2006; Capsoni et al. 2013). A similar action was also reported for the CD33 gene (Bradshaw et al. 2013). Also, macropinocytosis or phagocytosis is said to take up larger size Aβ aggregates (Mayor and Pagano 2007). Thus, by regulating the amount of aggregated Tau and Aβ protein, the prevalence of neurodegenerative diseases can be regulated (Fig. 4b).
Conclusion
In conclusion, Tau pathology and cholesterol metabolism appear to play a pivotal and primary role of neurodegeneration in AD and several other Tauopathies. Many studies have focused on cholesterol metabolism and its impact on amyloid-β protein. Now since Tau protein is considered equally important in the pathogenesis of neurodegenerative disease, especially Alzheimer's, it is important to know the molecular mechanisms involved in its hyperphosphorylation. As per the earlier reports, cholesterol turnover in the brain contributes majorly in regulating Tau phosphorylation and pathogenesis. The article provides an overview of the molecular mechanism involving microglia and the genes in controlling the concentration of APOE-containing cholesterol and Tau protein. Hence, to the best of our knowledge the review article provides a strong background to fully elucidate the role of microglia in modulating the Tau pathology and cholesterol metabolism in the brain.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-β
- FAD:
-
Familial Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- PS-1 & 2:
-
Presenilin-1 & 2
- CNS:
-
Central nervous system
- BBB:
-
Blood–brain barrier
- HMG-CoA:
-
3-Hydroxy-3-methylglutaryl-CoA
- ABCA1:
-
ATP-binding cassette transporter
- CERP:
-
Cholesterol efflux regulatory protein
- NPC:
-
Niemann–Pick type C
- ACAT1/SOAT1:
-
Acyl-coenzyme A: cholesterol acyltransferase-1
- 24- OHC:
-
24-Hydroxycholesterol
- LXR:
-
Liver X receptor
- CaMKII:
-
Calcium and calmodulin-dependent protein kinase- II
- TLR4:
-
Toll-like receptor-4
- CX3CR1:
-
CX3C chemokine receptor-1
- PI3K:
-
Phosphoinositide 3-kinase
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- SORL1:
-
Sortilin-related receptor 1
- ER:
-
Endoplasmic reticulum
- APOE:
-
Apolipoprotein E
- SREBPs:
-
Sterol regulatory element-binding proteins
- LDL:
-
Low-density lipoprotein
- VLDLR:
-
Very low-density lipoprotein receptor
- LRP1:
-
LDL receptor-related protein 1
- GWAS:
-
Genome-wide association studies
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- RCT:
-
Reverse cholesterol transport
- MAP:
-
Microtubule-associated protein
- PHFs:
-
Paired helical filaments
- GSK-3:
-
Glycogen synthase kinase-3
- cdk5:
-
Cyclin-dependent protein kinase-5
- PKA:
-
Protein kinase A
- SIGLEC:
-
Sialic acid-binding immunoglobulin-type lectins
- BIN1:
-
Bridging integrator 1
- CME:
-
Clathrin-mediated endocytosis
- LOAD:
-
Late-onset AD
- HSPG:
-
Heparin sulfate proteoglycan
- FAK:
-
Focal adhesion kinase
References
Abraha A, Ghoshal N, Gamblin TC et al (2000) C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J Cell Sci 113(Pt 21):3737–3745
Akram A, Schmeidler J, Katsel P et al (2010) Increased expression of cholesterol transporter ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain Res 1318:167–177. https://doi.org/10.1016/j.brainres.2010.01.006
del Alonso AC, Mederlyova A, Novak M et al (2004) Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem 279:34873–34881. https://doi.org/10.1074/jbc.M405131200
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA 91:5562–5566
Andersen OM, Reiche J, Schmidt V et al (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA 102:13461–13466. https://doi.org/10.1073/pnas.0503689102
Andersson M, Elmberger PG, Edlund C et al (1990) Rates of cholesterol, ubiquinone, dolichol and dolichyl-P biosynthesis in rat brain slices. FEBS Lett 269:15–18
Andreadis A (2005) Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta 1739:91–103. https://doi.org/10.1016/j.bbadis.2004.08.010
Arendt T, Stieler JT, Holzer M (2016) Tau and tauopathies. Brain Res Bull 126:238–292. https://doi.org/10.1016/j.brainresbull.2016.08.018
Ashford JW (2004) APOE genotype effects on Alzheimer’s disease onset and epidemiology. J Mol Neurosci 23:157–165. https://doi.org/10.1385/JMN:23:3:157
Avila J, Jiménez JS, Sayas CL et al (2016) Tau structures. Front Aging Neurosci 8:262. https://doi.org/10.3389/fnagi.2016.00262
Bae SH, Lee JN, Fitzky BU et al (1999) Cholesterol biosynthesis from lanosterol. Molecular cloning, tissue distribution, expression, chromosomal localization, and regulation of rat 7-dehydrocholesterol reductase, a Smith-Lemli-Opitz syndrome-related protein. J Biol Chem 274:14624–14631
Ballatore C, Lee VM-Y, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8:663–672. https://doi.org/10.1038/nrn2194
Bao M, Hanabuchi S, Facchinetti V et al (2012) CD2AP/SHIP1 complex positively regulates plasmacytoid dendritic cell receptor signaling by inhibiting the E3 ubiquitin ligase Cbl. J Immunol 189:786–792. https://doi.org/10.4049/jimmunol.1200887
Baranowski M (2008) Biological role of liver X receptors. J Physiol Pharmacol 59(Suppl 7):31–55
Bertram L, Tanzi RE (2005) The genetic epidemiology of neurodegenerative disease. J Clin Investig 115:1449–1457. https://doi.org/10.1172/JCI24761
Bhaskar K, Konerth M, Kokiko-Cochran ON et al (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68:19–31. https://doi.org/10.1016/j.neuron.2010.08.023
Björkhem I, Heverin M, Leoni V et al (2006) Oxysterols and Alzheimer’s disease. Acta Neurol Scand Suppl 185:43–49. https://doi.org/10.1111/j.1600-0404.2006.00684.x
Björkhem I, Meaney S (2004) Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24:806–815. https://doi.org/10.1161/01.ATV.0000120374.59826.1b
Block RC, Dorsey ER, Beck CA et al (2010) Altered cholesterol and fatty acid metabolism in Huntington disease. J Clin Lipidol 4:17–23. https://doi.org/10.1016/j.jacl.2009.11.003
Bolós M, Llorens-Martín M, Jurado-Arjona J et al (2016) Direct evidence of internalization of Tau by microglia in vitro and in vivo. J Alzheimers Dis 50:77–87. https://doi.org/10.3233/JAD-150704
Bolós M, Llorens-Martín M, Perea JR et al (2017a) Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener 12:59. https://doi.org/10.1186/s13024-017-0200-1
Bolós M, Pallas-Bazarra N, Terreros-Roncal J et al (2017b) Soluble Tau has devastating effects on the structural plasticity of hippocampal granule neurons. Transl Psychiatry 7:1267. https://doi.org/10.1038/s41398-017-0013-6
Boucrot E, Ferreira APA, Almeida-Souza L et al (2015) Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 517:460–465. https://doi.org/10.1038/nature14067
Bradshaw EM, Chibnik LB, Keenan BT et al (2013) CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci 16:848–850. https://doi.org/10.1038/nn.3435
Brelstaff J, Tolkovsky AM, Ghetti B et al (2018) Living neurons with Tau filaments aberrantly expose phosphatidylserine and are phagocytosed by microglia. Cell Rep 24:1939–1948.e4. https://doi.org/10.1016/j.celrep.2018.07.072
Brier MR, Gordon B, Friedrichsen K et al (2016) Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med 8:338ra66. https://doi.org/10.1126/scitranslmed.aaf2362
Bryleva EY, Rogers MA, Chang CCY et al (2010) ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci USA 107:3081–3086. https://doi.org/10.1073/pnas.0913828107
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344. https://doi.org/10.1038/nrn2620
Busche MA, Wegmann S, Dujardin S et al (2019) Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat Neurosci. https://doi.org/10.1038/s41593-018-0289-8
Calafate S, Flavin W, Verstreken P, Moechars D (2016) Loss of Bin1 promotes the propagation of Tau pathology. Cell Rep 17:931–940. https://doi.org/10.1016/j.celrep.2016.09.063
Capsoni S, Carlo A-S, Vignone D et al (2013) SorLA deficiency dissects amyloid pathology from tau and cholinergic neurodegeneration in a mouse model of Alzheimer’s disease. J Alzheimers Dis 33:357–371. https://doi.org/10.3233/JAD-2012-121399
Carbajosa G, Malki K, Lawless N et al (2018) Loss of Trem2 in microglia leads to widespread disruption of cell coexpression networks in mouse brain. Neurobiol Aging 69:151–166. https://doi.org/10.1016/j.neurobiolaging.2018.04.019
Carrasquillo MM, Belbin O, Hunter TA et al (2010) Replication of CLU, CR1, and PICALM associations with alzheimer disease. Arch Neurol 67:961–964. https://doi.org/10.1001/archneurol.2010.147
Carstea ED, Morris JA, Coleman KG et al (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277:228–231
Chapuis J, Hansmannel F, Gistelinck M et al (2013) Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 18:1225–1234. https://doi.org/10.1038/mp.2013.1
Cho S-H, Sun B, Zhou Y et al (2011) CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem 286:32713–32722. https://doi.org/10.1074/jbc.M111.254268
Christianson HC, Belting M (2014) Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol 35:51–55. https://doi.org/10.1016/j.matbio.2013.10.004
Colonna M, Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 35:441–468. https://doi.org/10.1146/annurev-immunol-051116-052358
Corder EH, Saunders AM, Strittmatter WJ et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Couerbe JP (1984) Du cerveau, considéré sous le point du vue chimique et physiologique. Ann Chim Phys 56:160–193
Cummings JL (2004) Alzheimer’s disease. N Engl J Med 351:56–67. https://doi.org/10.1056/NEJMra040223
Das R, Balmik AA, Chinnathambi S (2020) Phagocytosis of full-length Tau oligomers by Actin-remodeling of activated microglia. J Neuroinflamm 17:10. https://doi.org/10.1186/s12974-019-1694-y
de Chaves EI, Rusiñol AE, Vance DE et al (1997) Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem 272:30766–30773
Deane R, Wu Z, Sagare A et al (2004) LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43:333–344. https://doi.org/10.1016/j.neuron.2004.07.017
Dedieu S, Langlois B (2008) LRP-1: a new modulator of cytoskeleton dynamics and adhesive complex turnover in cancer cells. Cell Adh Migr 2:77–80
DeGrella RF, Simoni RD (1982) Intracellular transport of cholesterol to the plasma membrane. J Biol Chem 257:14256–14262
Di Paolo G, Kim T-W (2011) Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci 12:284–296. https://doi.org/10.1038/nrn3012
Dietschy JM (2009) Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem 390:287–293. https://doi.org/10.1515/BC.2009.035
Dietschy JM, Turley SD (2001) Cholesterol metabolism in the brain. Curr Opin Lipidol 12:105–112
Dietschy JM, Turley SD (2004) Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 45:1375–1397. https://doi.org/10.1194/jlr.R400004-JLR200
Duthey B (2013) Background paper 6.11: Alzheimer disease and other dementias. A public health approach to innovation
Eberlé D, Hegarty B, Bossard P et al (2004) SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86:839–848. https://doi.org/10.1016/j.biochi.2004.09.018
Elali A, Rivest S (2013) The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer’s disease. Front Physiol 4:45. https://doi.org/10.3389/fphys.2013.00045
Ellis JD, Barrios-Rodiles M, Colak R et al (2012) Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell 46:884–892. https://doi.org/10.1016/j.molcel.2012.05.037
Fan Q-W, Yu W, Senda T et al (2001) Cholesterol-dependent modulation of tau phosphorylation in cultured neurons. J Neurochem 76:391–400. https://doi.org/10.1046/j.1471-4159.2001.00063.x
Fester L, Zhou L, Bütow A et al (2009) Cholesterol-promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus 19:692–705. https://doi.org/10.1002/hipo.20548
Fjorback AW, Seaman M, Gustafsen C et al (2012) Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci 32:1467–1480. https://doi.org/10.1523/JNEUROSCI.2272-11.2012
Gabbi C, Warner M, Gustafsson J-Å (2014) Action mechanisms of Liver X Receptors. Biochem Biophys Res Commun 446:647–650. https://doi.org/10.1016/j.bbrc.2013.11.077
Gamba P, Testa G, Gargiulo S et al (2015) Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2015.00119
Gaylor JL (2002) Membrane-bound enzymes of cholesterol synthesis from lanosterol. Biochem Biophys Res Commun 292:1139–1146. https://doi.org/10.1006/bbrc.2001.2008
Glöckner F, Meske V, Lütjohann D, Ohm TG (2011) Dietary cholesterol and its effect on tau protein: a study in apolipoprotein e-deficient and P301L human tau mice. J Neuropathol Exp Neurol 70:292–301. https://doi.org/10.1097/NEN.0b013e318212f185
Glöckner F, Ohm TG (2014) Tau pathology induces intraneuronal cholesterol accumulation. J Neuropathol Exp Neurol 73:846–854. https://doi.org/10.1097/NEN.0000000000000103
Goedert M, Spillantini MG (2011) Pathogenesis of the tauopathies. J Mol Neurosci 45:425–431. https://doi.org/10.1007/s12031-011-9593-4
Gong C-X, Iqbal K (2008) Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 15:2321–2328
Gong C-X, Liu F, Grundke-Iqbal I, Iqbal K (2006) Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimers Dis 9:1–12
Gosselet F, Candela P, Sevin E et al (2009) Transcriptional profiles of receptors and transporters involved in brain cholesterol homeostasis at the blood-brain barrier: use of an in vitro model. Brain Res 1249:34–42. https://doi.org/10.1016/j.brainres.2008.10.036
Griciuc A, Serrano-Pozo A, Parrado AR et al (2013) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78:631–643. https://doi.org/10.1016/j.neuron.2013.04.014
Guerreiro R, Wojtas A, Bras J et al (2013a) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127. https://doi.org/10.1056/NEJMoa1211851
Guerreiro R, Wojtas A, Bras J et al (2013b) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127. https://doi.org/10.1056/NEJMoa1211851
Haase C, Stieler JT, Arendt T, Holzer M (2004) Pseudophosphorylation of tau protein alters its ability for self-aggregation. J Neurochem 88:1509–1520
Hansen DV, Hanson JE, Sheng M (2017) Microglia in Alzheimer’s disease. J Cell Biol 217:459–472. https://doi.org/10.3389/fphar.2012.00014
Heino S, Lusa S, Somerharju P et al (2000) Dissecting the role of the golgi complex and lipid rafts in biosynthetic transport of cholesterol to the cell surface. Proc Natl Acad Sci USA 97:8375–8380. https://doi.org/10.1073/pnas.140218797
Heneka MT, Golenbock DT, Latz E (2015) Innate immunity in Alzheimer’s disease. Nat Immunol 16:229–236. https://doi.org/10.1038/ni.3102
Heneka MT, Kummer MP, Stutz A et al (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678. https://doi.org/10.1038/nature11729
Ho L, Qin W, Pompl PN et al (2004) Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J 18:902–904. https://doi.org/10.1096/fj.03-0978fje
Holler CJ, Davis PR, Beckett TL et al (2014) Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J Alzheimers Dis 42:1221–1227. https://doi.org/10.3233/JAD-132450
Hollingworth P, Harold D, Sims R et al (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43:429–435. https://doi.org/10.1038/ng.803
Holtzman DM, Herz J, Bu G (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006312. https://doi.org/10.1101/cshperspect.a006312
Hooper C, Killick R, Lovestone S (2008) The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 104:1433–1439. https://doi.org/10.1111/j.1471-4159.2007.05194.x
Hopp SC, Lin Y, Oakley D et al (2018) The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflamm 15:269. https://doi.org/10.1186/s12974-018-1309-z
Hoyer S (2004) Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv Exp Med Biol 541:135–152
Hu W, Zhang X, Tung YC et al (2016) Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement 12:1066–1077. https://doi.org/10.1016/j.jalz.2016.01.014
Hutter-Paier B, Huttunen HJ, Puglielli L et al (2004) The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 44:227–238. https://doi.org/10.1016/j.neuron.2004.08.043
Iliff JJ, Nedergaard M (2013) Is there a cerebral lymphatic system? Stroke 44:S93–S95. https://doi.org/10.1161/STROKEAHA.112.678698
Iliff JJ, Wang M, Liao Y et al (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4:147ra111. https://doi.org/10.1126/scitranslmed.3003748
Iqbal K, del Alonso AC, Chen S, et al (2005a) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739:198–210. https://doi.org/10.1016/j.bbadis.2004.09.008
Iqbal K, Del C, Alonso A, Chen S et al (2005b) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739:198–210. https://doi.org/10.1016/j.bbadis.2004.09.008
Iqbal K, Liu F, Gong C-X et al (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 118:53–69. https://doi.org/10.1007/s00401-009-0486-3
Jay TR, von Saucken VE, Landreth GE (2017) TREM2 in neurodegenerative diseases. Mol Neurodegener 12:56. https://doi.org/10.1186/s13024-017-0197-5
Johnson GV, Hartigan JA (1999) Tau protein in normal and Alzheimer’s disease brain: an update. J Alzheimers Dis 1:329–351
Jolivalt CG, Lee CA, Beiswenger KK et al (2008) Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res 86:3265–3274. https://doi.org/10.1002/jnr.21787
Jonsson T, Stefansson H, Steinberg S et al (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368:107–116. https://doi.org/10.1056/NEJMoa1211103
Kajiho H, Sakurai K, Minoda T et al (2011) Characterization of RIN3 as a guanine nucleotide exchange factor for the Rab5 subfamily GTPase Rab31. J Biol Chem 286:24364–24373. https://doi.org/10.1074/jbc.M110.172445
Kallen CB, Billheimer JT, Summers SA et al (1998) Steroidogenic Acute Regulatory Protein (StAR) is a sterol transfer protein. J Biol Chem 273:26285–26288. https://doi.org/10.1074/jbc.273.41.26285
Kanekiyo T, Bu G (2014) The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2014.00093
Kanekiyo T, Cirrito JR, Liu C-C et al (2013) Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci 33:19276–19283. https://doi.org/10.1523/JNEUROSCI.3487-13.2013
Kanekiyo T, Zhang J, Liu Q et al (2011) Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci 31:1644–1651. https://doi.org/10.1523/JNEUROSCI.5491-10.2011
Kang J, Rivest S (2012) Lipid metabolism and neuroinflammation in Alzheimer’s disease: a role for liver X receptors. Endocr Rev 33:715–746. https://doi.org/10.1210/er.2011-1049
Kaplan MR, Simoni RD (1985) Transport of cholesterol from the endoplasmic reticulum to the plasma membrane. J Cell Biol 101:446–453
Karch CM, Cruchaga C, Goate AM (2014) Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83:11–26. https://doi.org/10.1016/j.neuron.2014.05.041
Karch CM, Goate AM (2015) Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77:43–51. https://doi.org/10.1016/j.biopsych.2014.05.006
Karten B, Campenot RB, Vance DE, Vance JE (2006) Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J Biol Chem 281:4049–4057. https://doi.org/10.1074/jbc.M508915200
Ke YD, Delerue F, Gladbach A et al (2009) Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 4:e7917. https://doi.org/10.1371/journal.pone.0007917
Khatoon S, Grundke-Iqbal I, Iqbal K (1992) Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem 59:750–753
Kim K-W, Vallon-Eberhard A, Zigmond E et al (2011) In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118:e156–e167. https://doi.org/10.1182/blood-2011-04-348946
Kim WS, Weickert CS, Garner B (2008) Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem 104:1145–1166. https://doi.org/10.1111/j.1471-4159.2007.05099.x
Kitazawa M, Cheng D, Tsukamoto MR et al (2011) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol 187:6539–6549. https://doi.org/10.4049/jimmunol.1100620
Kitazawa M, Oddo S, Yamasaki TR et al (2005) Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci 25:8843–8853. https://doi.org/10.1523/JNEUROSCI.2868-05.2005
Köpke E, Tung YC, Shaikh S et al (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 268:24374–24384
Korach-André M, Gustafsson J-Å (2015) Liver X receptors as regulators of metabolism. Biomol Concepts 6:177–190. https://doi.org/10.1515/bmc-2015-0007
Korade Z, Kenworthy AK (2008) Lipid rafts, cholesterol, and the brain. Neuropharmacology 55:1265–1273. https://doi.org/10.1016/j.neuropharm.2008.02.019
Koronyo-Hamaoui M, Ko MK, Koronyo Y et al (2009) Attenuation of AD-like neuropathology by harnessing peripheral immune cells: local elevation of IL-10 and MMP-9. J Neurochem 111:1409–1424. https://doi.org/10.1111/j.1471-4159.2009.06402.x
Lahiri DK (2004) Apolipoprotein E as a target for developing new therapeutics for Alzheimer’s disease based on studies from protein, RNA, and regulatory region of the gene. J Mol Neurosci 23:225–233. https://doi.org/10.1385/JMN:23:3:225
Lambert JC, Ibrahim-Verbaas CA, Harold D et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45:1452–1458. https://doi.org/10.1038/ng.2802
Lawson LJ, Perry VH, Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48:405–415. https://doi.org/10.1016/0306-4522(92)90500-2
Leoni V, Caccia C (2015) The impairment of cholesterol metabolism in Huntington disease. Biochim Biophys Acta 1851:1095–1105. https://doi.org/10.1016/j.bbalip.2014.12.018
Li J-T, Zhang Y (2018) TREM2 regulates innate immunity in Alzheimer’s disease. J Neuroinflamm 15:107. https://doi.org/10.1186/s12974-018-1148-y
Li J, Kanekiyo T, Shinohara M et al (2012) Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem 287:44593–44601. https://doi.org/10.1074/jbc.M112.420224
Li Y, Lu W, Marzolo MP, Bu G (2001) Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J Biol Chem 276:18000–18006. https://doi.org/10.1074/jbc.M101589200
Lindwall G, Cole RD (1984) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259:5301–5305
Linetti A, Fratangeli A, Taverna E et al (2010) Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci 123:595–605. https://doi.org/10.1242/jcs.060681
Litvinov DY, Savushkin EV, Dergunov AD (2018) Intracellular and plasma membrane events in cholesterol transport and homeostasis. J Lipids 2018:1–22. https://doi.org/10.1155/2018/3965054
Liu F, Gong C-X (2008) Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener 3:8. https://doi.org/10.1186/1750-1326-3-8
Liu F, Li B, Tung E-J et al (2007a) Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci 26:3429–3436. https://doi.org/10.1111/j.1460-9568.2007.05955.x
Liu Q, Trotter J, Zhang J et al (2010) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci 30:17068–17078. https://doi.org/10.1523/JNEUROSCI.4067-10.2010
Liu Q, Zerbinatti CV, Zhang J et al (2007b) Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56:66–78. https://doi.org/10.1016/j.neuron.2007.08.008
Lizcano JM, Alessi DR (2002) The insulin signalling pathway. Curr Biol 12:R236–R238
Luo W, Liu W, Hu X et al (2015) Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 5:11161. https://doi.org/10.1038/srep11161
Ma Y, Yang H, Qi J et al (2010) CD2AP is indispensable to multistep cytotoxic process by NK cells. Mol Immunol 47:1074–1082. https://doi.org/10.1016/j.molimm.2009.11.004
Maccioni RB, Farías G, Morales I, Navarrete L (2010) The revitalized tau hypothesis on Alzheimer’s disease. Arch Med Res 41:226–231. https://doi.org/10.1016/j.arcmed.2010.03.007
Maher PA, Schubert DR (2009) Metabolic links between diabetes and Alzheimer’s disease. Expert Rev Neurother 9:617–630. https://doi.org/10.1586/ern.09.18
Mahley RW (2016) Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med 94:739–746
Mahley RW, Weisgraber KH, Huang Y (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci USA 103:5644–5651. https://doi.org/10.1073/pnas.0600549103
Malik M, Parikh I, Vasquez JB et al (2015) Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol Neurodegener 10:52. https://doi.org/10.1186/s13024-015-0048-1
Malik M, Simpson JF, Parikh I et al (2013) CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 33:13320–13325. https://doi.org/10.1523/JNEUROSCI.1224-13.2013
Manna P, Jain SK (2015) Phosphatidylinositol-3,4,5-triphosphate and cellular signaling: implications for obesity and diabetes. Cell Physiol Biochem 35:1253–1275. https://doi.org/10.1159/000373949
Manolopoulos KN, Klotz L-O, Korsten P et al (2010) Linking Alzheimer’s disease to insulin resistance: the FoxO response to oxidative stress. Mol Psychiatry 15:1046–1052. https://doi.org/10.1038/mp.2010.17
Mantuano E, Jo M, Gonias SL, Campana WM (2010) Low density lipoprotein receptor-related protein (LRP1) regulates Rac1 and RhoA reciprocally to control Schwann cell adhesion and migration. J Biol Chem 285:14259–14266. https://doi.org/10.1074/jbc.M109.085126
Maphis N, Xu G, Kokiko-Cochran ON et al (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138:1738–1755. https://doi.org/10.1093/brain/awv081
Matsuzaki T, Sasaki K, Tanizaki Y et al (2010) Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 75:764–770. https://doi.org/10.1212/WNL.0b013e3181eee25f
Mayor S, Pagano RE (2007) Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol 8:603–612. https://doi.org/10.1038/nrm2216
Mazaheri F, Snaidero N, Kleinberger G et al (2017) TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep 18:1186–1198. https://doi.org/10.15252/embr.201743922
McMillin M, DeMorrow S (2016) Effects of bile acids on neurological function and disease. FASEB J 30:3658–3668. https://doi.org/10.1096/fj.201600275R
Medeiros R, Baglietto-Vargas D, LaFerla FM (2011) The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther 17:514–524. https://doi.org/10.1111/j.1755-5949.2010.00177.x
Medina M, Avila J (2014) The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci 8:113. https://doi.org/10.3389/fncel.2014.00113
Michikawa M (2006) Role of cholesterol in amyloid cascade: cholesterol-dependent modulation of tau phosphorylation and mitochondrial function. Acta Neurol Scand Suppl 185:21–26. https://doi.org/10.1111/j.1600-0404.2006.00681.x
Minagawa H, Gong J-S, Jung C-G et al (2009) Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J Neurosci Res 87:2498–2508. https://doi.org/10.1002/jnr.22073
Morell P, Jurevics H (1996) Origin of cholesterol in myelin. Neurochem Res 21:463–470
Morris M, Knudsen GM, Maeda S et al (2015) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci 18:1183–1189. https://doi.org/10.1038/nn.4067
Naj AC, Jun G, Beecham GW et al (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436–441. https://doi.org/10.1038/ng.801
Nayak D, Roth TL, McGavern DB (2014) Microglia development and function. Annu Rev Immunol 32:367–402. https://doi.org/10.1146/annurev-immunol-032713-120240
Offe K, Dodson SE, Shoemaker JT et al (2006) The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci 26:1596–1603. https://doi.org/10.1523/JNEUROSCI.4946-05.2006
Orth M, Bellosta S (2012) Cholesterol: its regulation and role in central nervous system disorders. Cholesterol 2012:1–19. https://doi.org/10.1155/2012/292598
Orth M, Weng W, Funke H et al (1999) Effects of a frequent apolipoprotein E isoform, ApoE4Freiburg (Leu28–%3ePro), on lipoproteins and the prevalence of coronary artery disease in whites. Arterioscler Thromb Vasc Biol 19:1306–1315
Paolicelli RC, Bolasco G, Pagani F et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458. https://doi.org/10.1126/science.1202529
Pei J-J, Gong C-X, An W-L et al (2003) Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer’s disease. Am J Pathol 163:845–858. https://doi.org/10.1016/S0002-9440(10)63445-1
Peng Q, Malhotra S, Torchia JA et al (2010) TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal 3:ra38. https://doi.org/10.1126/scisignal.2000500
Perea JR, Llorens-Martín M, Ávila J, Bolós M (2018) The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front Cell Neurosci 12:1–8. https://doi.org/10.3389/fncel.2018.00172
Pereira JB, Ossenkoppele R, Palmqvist S et al (2019) Amyloid and tau accumulate across distinct spatial networks and are differentially associated with brain connectivity. Elife 8:e50830. https://doi.org/10.7554/eLife.50830
Pérez-Cerdá F, Sánchez-Gómez MV, Matute C (2015) Pío del Río Hortega and the discovery of the oligodendrocytes. Front Neuroanat. https://doi.org/10.3389/fnana.2015.00092
Pfrieger FW, Ungerer N (2011) Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 50:357–371. https://doi.org/10.1016/j.plipres.2011.06.002
Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423:435–439
Poppek D, Keck S, Ermak G et al (2006) Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem J 400:511–520. https://doi.org/10.1042/BJ20060463
Pottier C, Hannequin D, Coutant S et al (2012) High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry 17:875–879. https://doi.org/10.1038/mp.2012.15
Prokic I, Cowling BS, Laporte J (2014) Amphiphysin 2 (BIN1) in physiology and diseases. J Mol Med (Berl) 92:453–463. https://doi.org/10.1007/s00109-014-1138-1
Puglielli L, Tanzi RE, Kovacs DM (2003) Alzheimer’s disease: the cholesterol connection. Nat Neurosci 6:345–351. https://doi.org/10.1038/nn0403-345
Qing-Qing T, Yu-Chao C, Zhi-Ying W (2018) The role of CD2AP in the pathogenesis of Alzheimer’s Disease. Aging Dis. https://doi.org/10.14336/AD.2018.1025
Qu Z, Jiao Z, Sun X et al (2011) Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res 1383:300–306. https://doi.org/10.1016/j.brainres.2011.01.084
Radhakrishnan A, Ikeda Y, Kwon HJ et al (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A 104:6511–6518. https://doi.org/10.1073/pnas.0700899104
Rahman A, Akterin S, Flores-Morales A et al (2005) High cholesterol diet induces tau hyperphosphorylation in apolipoprotein E deficient mice. FEBS Lett 579:6411–6416. https://doi.org/10.1016/j.febslet.2005.10.024
Raj T, Ryan KJ, Replogle JM et al (2014) CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet 23:2729–2736. https://doi.org/10.1093/hmg/ddt666
Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV (2015) Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front Aging Neurosci 7:136. https://doi.org/10.3389/fnagi.2015.00136
Ramirez DMO, Andersson S, Russell DW (2008) Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol 507:1676–1693. https://doi.org/10.1002/cne.21605
Ramjaun AR, McPherson PS (1998) Multiple amphiphysin II splice variants display differential clathrin binding: identification of two distinct clathrin-binding sites. J Neurochem 70:2369–2376
Rankin CA, Sun Q, Gamblin TC (2007) Tau phosphorylation by GSK-3β promotes tangle-like filament morphology. Mol Neurodegener 2:12. https://doi.org/10.1186/1750-1326-2-12
Rebeck GW, Reiter JS, Strickland DK, Hyman BT (1993) Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 11:575–580
Reid PC, Sakashita N, Sugii S et al (2004) A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J Lipid Res 45:582–591. https://doi.org/10.1194/jlr.D300032-JLR200
Rodrıguez-Rodrıguez E, Vázquez-Higuera JL, Sánchez-Juan P et al (2010) Epistasis between intracellular cholesterol trafficking-related genes (NPC1 and ABCA1) and Alzheimer’s disease risk. J Alzheimer’s Dis 21:619–625. https://doi.org/10.3233/JAD-2010-100432
Rogers MA, Liu J, Song B-L et al (2015) Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. J Steroid Biochem Mol Biol 151:102–107. https://doi.org/10.1016/j.jsbmb.2014.09.008
Rouka E, Simister PC, Janning M et al (2015) Differential recognition preferences of the three Src homology 3 (SH3) domains from the adaptor CD2-associated protein (CD2AP) and direct association with Ras and Rab interactor 3 (RIN3). J Biol Chem 290:25275–25292. https://doi.org/10.1074/jbc.M115.637207
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806. https://doi.org/10.1038/414799a
Sato N, Morishita R (2014) Brain alterations and clinical symptoms of dementia in diabetes: aβ/tau-dependent and independent mechanisms. Front Endocrinol (Lausanne) 5:143. https://doi.org/10.3389/fendo.2014.00143
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8:595–608. https://doi.org/10.15252/emmm.201606210
Sengupta A, Kabat J, Novak M et al (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys 357:299–309. https://doi.org/10.1006/abbi.1998.0813
Shibata M, Yamada S, Kumar SR et al (2000) Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Investig 106:1489–1499. https://doi.org/10.1172/JCI10498
Shulman JM, Imboywa S, Giagtzoglou N et al (2014) Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum Mol Genet 23:870–877. https://doi.org/10.1093/hmg/ddt478
Simard AR, Soulet D, Gowing G et al (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49:489–502. https://doi.org/10.1016/j.neuron.2006.01.022
Simón D, Hernández F, Avila J (2013) The involvement of cholinergic neurons in the spreading of tau pathology. Front Neurol 4:74. https://doi.org/10.3389/fneur.2013.00074
Singh TJ, Grundke-Iqbal I, McDonald B, Iqbal K (1994) Comparison of the phosphorylation of microtubule-associated protein tau by non-proline dependent protein kinases. Mol Cell Biochem 131:181–189
Slepnev VI, Ochoa GC, Butler MH, De Camilli P (2000) Tandem arrangement of the clathrin and AP-2 binding domains in amphiphysin 1 and disruption of clathrin coat function by amphiphysin fragments comprising these sites. J Biol Chem 275:17583–17589. https://doi.org/10.1074/jbc.M910430199
Soccio RE, Breslow JL (2004) Intracellular cholesterol transport. Arterioscler Thromb Vasc Biol 24:1150–1160. https://doi.org/10.1161/01.ATV.0000131264.66417.d5
Sonawane SK, Chinnathambi S (2018) Prion-Like propagation of post-translationally modified tau in Alzheimer’s disease: a hypothesis. J Mol Neurosci 65:480–490. https://doi.org/10.1007/s12031-018-1111-5
Song H-L, Shim S, Kim D-H et al (2014) β-Amyloid is transmitted via neuronal connections along axonal membranes. Ann Neurol 75:88–97. https://doi.org/10.1002/ana.24029
Spuch C, Ortolano S, Navarro C (2012) LRP-1 and LRP-2 receptors function in the membrane neuron. Trafficking mechanisms and proteolytic processing in Alzheimer’s disease. Front Physiol 3:269. https://doi.org/10.3389/fphys.2012.00269
Starks EJ, Patrick O’Grady J, Hoscheidt SM et al (2015) Insulin resistance is associated with higher cerebrospinal fluid Tau levels in asymptomatic APOEɛ4 carriers. J Alzheimers Dis 46:525–533. https://doi.org/10.3233/JAD-150072
Swathi P (2013) A deeper look into cholesterol synthesis. In: ASBMB Today. https://www.asbmb.org/asbmbtoday/asbmbtoday_article.aspx?id=48189
Szaruga M, Munteanu B, Lismont S et al (2017) Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell 170:443–456.e14. https://doi.org/10.1016/j.cell.2017.07.004
Takei Y, Teng J, Harada A, Hirokawa N (2000) Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol 150:989–1000
Tan M-S, Yu J-T, Tan L (2013) Bridging integrator 1 (BIN1): form, function, and Alzheimer’s disease. Trends Mol Med 19:594–603. https://doi.org/10.1016/j.molmed.2013.06.004
Tarasoff-Conway JM, Carare RO, Osorio RS et al (2015) Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 11:457–470. https://doi.org/10.1038/nrneurol.2015.119
Tarr PT, Edwards PA (2008) ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP-2. J Lipid Res 49:169–182. https://doi.org/10.1194/jlr.M700364-JLR200
Teng J, Takei Y, Harada A et al (2001) Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J Cell Biol 155:65–76. https://doi.org/10.1083/jcb.200106025
Terwel D, Steffensen KR, Verghese PB et al (2011) Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J Neurosci 31:7049–7059. https://doi.org/10.1523/JNEUROSCI.6546-10.2011
Tian Y, Chang JC, Fan EY et al (2013) Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci USA 110:17071–17076. https://doi.org/10.1073/pnas.1315110110
Ulrich JD, Ulland TK, Colonna M, Holtzman DM (2017) Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 94:237–248. https://doi.org/10.1016/j.neuron.2017.02.042
van der Kant R, Langness VF, Herrera CM et al (2019) Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 24:363–375.e9. https://doi.org/10.1016/j.stem.2018.12.013
Vanier MT (2015) Complex lipid trafficking in Niemann-Pick disease type C. J Inherit Metab Dis 38:187–199. https://doi.org/10.1007/s10545-014-9794-4
Wang JZ, Grundke-Iqbal I, Iqbal K (1996) Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res 38:200–208
Wang Q, Yan J, Chen X et al (2011) Statins: multiple neuroprotective mechanisms in neurodegenerative diseases. Exp Neurol 230:27–34. https://doi.org/10.1016/j.expneurol.2010.04.006
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:22–35. https://doi.org/10.1038/nrn.2015.1
Wang Y, Rogers PM, Su C et al (2008) Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J Biol Chem 283:26332–26339. https://doi.org/10.1074/jbc.M804808200
Wegmann S, Jung YJ, Chinnathambi S et al (2010) Human Tau isoforms assemble into ribbon-like fibrils that display polymorphic structure and stability. J Biol Chem 285:27302–27313. https://doi.org/10.1074/jbc.M110.145318
Weller RO, Subash M, Preston SD et al (2008) Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol 18:253–266. https://doi.org/10.1111/j.1750-3639.2008.00133.x
Wildsmith KR, Holley M, Savage JC et al (2013) Evidence for impaired amyloid β clearance in Alzheimer’s disease. Alzheimers Res Ther 5:33. https://doi.org/10.1186/alzrt187
Wilsie LC, Orlando RA (2003) The low density lipoprotein receptor-related protein complexes with cell surface heparan sulfate proteoglycans to regulate proteoglycan-mediated lipoprotein catabolism. J Biol Chem 278:15758–15764. https://doi.org/10.1074/jbc.M208786200
Witzlack T, Wenzeck T, Thiery J, Orth M (2007) cAMP-induced expression of ABCA1 is associated with MAP-kinase-pathway activation. Biochem Biophys Res Commun 363:89–94. https://doi.org/10.1016/j.bbrc.2007.08.109
Wolf G, Stahl RA (2003) CD2-associated protein and glomerular disease. Lancet 362:1746–1748. https://doi.org/10.1016/S0140-6736(03)14856-8
Wolfe C, Fitz N, Nam K et al (2019) The Role of APOE and TREM2 in Alzheimer’s disease—current understanding and perspectives. Int J Mol Sci 20:81. https://doi.org/10.3390/ijms20010081
Wollmer MA, Streffer JR, Tsolaki M et al (2003) Genetic association of acyl-coenzyme A: cholesterol acyltransferase with cerebrospinal fluid cholesterol levels, brain amyloid load, and risk for Alzheimer’s disease. Mol Psychiatry 8:635–638. https://doi.org/10.1038/sj.mp.4001296
Wong BX, Hung YH, Bush AI, Duce JA (2014) Metals and cholesterol: two sides of the same coin in Alzheimer’s disease pathology. Front Aging Neurosci 6:91. https://doi.org/10.3389/fnagi.2014.00091
Wüstner D, Mondal M, Tabas I, Maxfield FR (2005) Direct observation of rapid internalization and intracellular transport of sterol by macrophage foam cells. Traffic 6:396–412. https://doi.org/10.1111/j.1600-0854.2005.00285.x
Xu S, Zhang X, Liu P (2018) Lipid droplet proteins and metabolic diseases. Biochim Biophys Acta 1864:1968–1983
Yeh FL, Wang Y, Tom I et al (2016) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91:328–340. https://doi.org/10.1016/j.neuron.2016.06.015
Yu C, Nwabuisi-Heath E, Laxton K, LaDu MJ (2010) Endocytic pathways mediating oligomeric Abeta42 neurotoxicity. Mol Neurodegener 5:19. https://doi.org/10.1186/1750-1326-5-19
Zefirov AL, Petrov AM (2010) Lipids in the process of synaptic vesicle exo- and endocytosis. Ross Fiziol zhurnal Im IM Sechenova 96:753–765
Zhan Y, Paolicelli RC, Sforazzini F et al (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17:400–406. https://doi.org/10.1038/nn.3641
Zhang J, Liu Q (2015) Cholesterol metabolism and homeostasis in the brain. Protein Cell 6:254–264. https://doi.org/10.1007/s13238-014-0131-3
Zheng H, Jia L, Liu C-C et al (2017) TREM2 promotes microglial survival by activating Wnt/β-catenin pathway. J Neurosci 37:1772–1784. https://doi.org/10.1523/JNEUROSCI.2459-16.2017
Zhou Y, Hayashi I, Wong J et al (2014) Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PLoS ONE 9:e103187. https://doi.org/10.1371/journal.pone.0103187
Acknowledgements
We are grateful to Chinnathambi lab members for their scientific discussions, helpful suggestions, and we highly appreciate the critical reading of the manuscript.
Funding
This project is supported in part by grant from in-house CSIR-National Chemical Laboratory grant MLP029526.
Author information
Authors and Affiliations
Contributions
SN and SC prepared the initial draft of the paper. HC and MC helped in preparing figure modifications and critical reading for the manuscript. SC conceived the idea of the work, supervised, resource provided, and wrote the paper. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Research Involving Human Participants and /or Animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nanjundaiah, S., Chidambaram, H., Chandrashekar, M. et al. Role of Microglia in Regulating Cholesterol and Tau Pathology in Alzheimer’s Disease. Cell Mol Neurobiol 41, 651–668 (2021). https://doi.org/10.1007/s10571-020-00883-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-020-00883-6