
Abstract
Neurodegeneration entails progressive loss of neuronal structure as well as function leading to cognitive failure, apathy, anxiety, irregular body movements, mood swing and ageing. Proteomic dysregulation is considered the key factor for neurodegeneration. Mechanisms involving deregulated processing of proteins such as amyloid beta (Aβ) oligomerization; tau hyperphosphorylation, prion misfolding; α-synuclein accumulation/lewy body formation, chaperone deregulation, acetylcholine depletion, adenosine 2A (A2A) receptor hyperactivation, secretase deregulation, leucine-rich repeat kinase 2 (LRRK2) mutation and mitochondrial proteinopathies have deeper implications in neurodegenerative disorders. Better understanding of such pathological mechanisms is pivotal for exploring crucial drug targets. Herein, we provide a comprehensive outlook about the diverse proteomic irregularities in Alzheimer’s, Parkinson’s and Creutzfeldt Jakob disease (CJD). We explicate the role of key neuroproteomic drug targets notably Aβ, tau, alpha synuclein, prions, secretases, acetylcholinesterase (AchE), LRRK2, molecular chaperones, A2A receptors, muscarinic acetylcholine receptors (mAchR), N-methyl-D-aspartate receptor (NMDAR), glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) and mitochondrial/oxidative stress-related proteins for combating neurodegeneration and associated cognitive and motor impairment. Cross talk between amyloidopathy, synucleinopathy, tauopathy and several other proteinopathies pinpoints the need to develop safe therapeutics with ability to strike multiple targets in the aetiology of the neurodegenerative disorders. Therapeutics like microtubule stabilisers, chaperones, kinase inhibitors, anti-aggregation agents and antibodies could serve promising regimens for treating neurodegeneration. However, drugs should be target specific, safe and able to penetrate blood–brain barrier.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Once degenerated, neurons of central nervous system (CNS) cannot replace themselves thus causing severe neurodegenerative disorders. The actual cause underlying regenerative failure of neurons is still not known. However, scientists report the presence of “road-blocks” in the injured or diseased CNS that prevent neurons from regenerating themselves. These road-blocks are diverse in nature and include molecules found in extracellular matrix as well as the proteins that get expressed once the neurons are damaged. In addition, once the CNS development is completed, there occurs activation of certain genes in neurons that act as brakes and prevent the further development of CNS. Nowadays, scientists are trying to silence such genes or their products so as to turn on growth programs in neurons.
Neurodegenerative diseases (ND’s) are growing at alarming pace due to rise in the elderly population (Heemels 2016). Some of these diseases damage person’s memory and cognition while others affect the ability to move, speak and breathe (Gitler et al. 2017). In America alone, around 5 million people suffer from Alzheimer’s disease (AD), 0.4 million from multiple sclerosis (MS), 1 million from Parkinson’s disease (PD), 30,000 from amyotrophic lateral sclerosis (ALS) and 3000 from Huntington’s disease (HD) (Agrawal and Biswas 2015). Globally, CJD affects about one person in every one million annually. In US alone, about 350 cases are detected annually. Cure for neurodegenerative diseases (NDs) is still under investigation and the current therapeutics only treat symptoms (Lang 2010). Necessity to develop effective drugs is the need of the hour; however, the task poses perplexing challenges owing to complex nature of NDs. Proteins act as important enzymes, biochemical regulators, electron transporters, signal transducers, receptors and scaffolding agents in diverse biological cascades. Their deregulated form can disrupt the normal cellular physiology and lead to NDs. Excessive accumulation of mutant or wild-type proteins with distorted conformations that aid aggregation is a characteristic feature of many neurodegenerative diseases including Parkinson’s, Alzheimer’s and CJD.
Strategy to strike proteomic drug targets represents an imperative approach for restricting neuronal damage. So far several drugs have been developed or being tested in various animal models (Fig. 1) for their neuroprotective action but the issues like (I) blood–brain barrier penetration, (II) toxicity and (III) limited drug efficacy often challenge the process. Conventional drug-discovery approaches based on one gene, one drug and one disease philosophy just modulate symptoms and do little to treat the root cause. Drugs that target only one protein are susceptible to drug resistance mainly due to the fact that even a single mutation in the target active site can substantially reduce the binding affinity of a drug and hence its efficacy. Drugs that strike multiple targets would require the unlikely event of concurrent mutations appearing in multiple protein targets. Thus, developing ‘magic shotgun drugs” instead of ‘magic bullet drugs” could play pivotal role in formulating effective regimens for neurodegenerative diseases and associated cognitive decline (Cornelis and Schyf 2011). This review provides mechanistic insight about diverse proteomic destabilisations that occur during neuronal damage and also elucidates potential proteomic drug targets (Fig. 2) to combat neurodegeneration.


Structures of drugs/molecules currently employed for the treatment of neurodegenerative diseases or being tested in clinical trials

Diverse proteins found deregulated during neurodegenerative diseases and the associated common pathological symptoms
Alzheimer’s Disease
Alzheimer’s disease (AD) is characterised by extreme dementia i.e. loss of overall cognitive functioning of brain like remembering, thinking or reasoning to such an extent that it interferes with person’s daily activities. It is the leading cause of death in elderly. At present, about 24 million people suffer from AD across the globe (Chakrabarti et al. 2015). As per the Alzheimer’s Association, about 5.4 million American people were affected by AD in 2016 (Rubenstein 2017). As per reports, by the year 2050, the population of AD patients in the United States will increase threefold over the number in 2000 (Hebert et al. 2003). Being an irreversible proteinopathy and a progressive brain disorder, it causes slow destruction of brain’s ability to memorise even simplest tasks. Unfortunately, there is no treatment to stop or reverse its progression, although some drugs may temporarily improve symptoms. AD is a complex neurological disorder primarily characterised by the accumulation of amyloid beta plaques (Aβ) in brain, but amyloidopathy is not necessarily the ultimate cause of Alzheimer’s disease. Infact, many other possible models have been proposed including N-APP-DR6-Caspase pathway, tauopathy, ApoE protein deregulation and environmental factors mediated proteinopathy, as illustrated below:
Amyloidopathy is considered the main cause of AD. Amyloid beta is not produced directly but indirectly from neuronal transmembrane amyloid precursor protein (APP). APP is encoded by app gene located on chromosome 21. APP protein is crucial for the growth, survival and post-injury repair of neuronal cells but its cleavage product amyloid beta (Aβ) generated proteolytically by the mutual action of two aspartyl proteases i.e. beta and gamma secretases leading to neurotoxicity and neuronal death (Koo et al. 1990). Extracellular cleavage of transmembrane APP glycoprotein by beta secretase generates a soluble extracellular fragment known as sAPPβ and a cell membrane-bound fragment called C99. Subsequent cleavage of C99 within its transmembrane domain by gamma secretase liberates an intracellular fragment called amino-terminal APP intracellular domain (AICD) and an extracellular fragment known as amyloid beta (Aβ) which is 40–42 amino acid long fibrils having molecular weight around 4kd. The later undergoes oligomerization to form abnormal clumps outside the neurons called Aβ plaques (Tiraboschi et al. 2004). These plaques trigger inflammatory response and oxidative stress in neurons thereby disrupting their normal communication and leading to their death (Bosco et al. 2011). Altogether these events constitute the amyloidogenic pathway of AD (Fig. 3). Formation of A oligomers can further activate microglia which in turn releases various neuroinflammatory cytokines that upregulate, and secretases increase APP levels and decrease A clearance in the brain thus causing further upsurge of A and subsequent formation of A oligomers and plaques in the brain (Alasmari et al. 2018). Amyloid beta plaques are of three types i.e. senile, diffuse and neuritic (D’Andrea and Nagele 2010). The diffuse plaques have a little effect on cognitive function, while the neuritic plaques are markedly involved in cognitive decline (Kokubo et al. 2009). It is hypothesised that diffused plaques appear during the early stages of Alzheimer’s disease while the neuritic plaques appear in the advanced stages. Further, studies show that clathrin and clathrin adaptor proteins involved in the endocytosis of APP can lead to increased intracellular levels of amyloid beta peptide, contributing to the progression of Alzheimer’s (Hallock et al. 2012).

Amyloidogenic pathway of Alzheimer’s disease: Beta secretase cleaves Amyloid precursor protein into soluble amyloid precursor protein-beta (APP-β) and C99 fragment. The later in the presence of gamma secretase cleaves to amyloid beta and AICD fragment. Amyloid beta Aβ undergoes oligomerization and plaque formation leading to inflammation, oxidative stress and finally neuronal death
Aβ is also involved in activation of apoptosis signal-regulating kinase-1 (ASK1) which is implicated in reactive oxygen species (ROS)-mediated activation of c-Jun N-terminal kinases (JNK) as well as apoptosis (Hashimoto et al. 2004; Kadowaki et al. 2005a, b). Thus, Aβ may indirectly lead to neuronal death via ASK1 activation. Amyloid beta also modulates redox factor-1 that plays crucial role in cell death signalling pathway and DNA repair by showing interaction with transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kappa B), Activator protein 1 (AP-1) and tumour protein p53 (Tan et al. 2009a, b). Amyloid beta may also induce oxidative damage, primarily through mitochondria also affects the cholesterol equilibrium, promoting neuronal toxicity due to generation of highly reactive free radical species (Harris et al. 1995; Colell et al. 2009). Overall, it seems Aβ plays crucial role in inducing neurotoxicity through mechanisms like triggering plaque formation, neuroinflammation, apoptosis as well as oxidative stress.
An updated version of amyloid hypothesis came in 2009 which suggests that amyloid beta protein may not be the real culprit of disease but a close relative of it namely N-amyloid precursor protein (N-APP). Accordingly, amyloid precursor protein (APP) is enzymatically cleaved by an unknown mechanism at its N terminal to generate N-terminal amyloid precursor protein (N-APP) fragment. This fragment triggers the self destructive pathway by binding to a neuronal receptor known as death receptor 6 (DR6). This receptor is expressed majorly in developing neurons of brain and is required for normal cell body death and axonal pruning both in vivo and after trophic factor deprivation in vitro. N-APP fragment acts as a ligand and interacts with DR6 causing neurodegeneration via activation of caspase-6 enzyme contributing to the pathology of Alzheimer’s disease (Nikolaev 2009). Therefore, N-APP/DR6/caspase 6 signalling pathway (Fig. 4) represents an important cascade implicated in AD pathophysiology that might be targeted and hijacked in the ageing AD brain. The strategy to either prevent the formation of N-APP fragment or block its interaction with death receptor 6 (DR6) may have better therapeutic outcome for AD.

Demonstration of N-APP-DR6-Caspase-6 pathway of Alzheimer’s disease AD. Beta secretase cleaves amyloid precursor protein (APP), generating two fragments. Then some unknown enzyme cleaves one of the fragments towards N terminal of APP to generate N- terminal fragment amyloid precursor protein known as N-APP fragment. The later binds to death receptor-6 (DR6) located in the neuronal membrane. N-APP/DR6 interaction triggers the apoptotic pathway by activating caspase-6 that causes neuronal death leading to Alzheimer’s disease
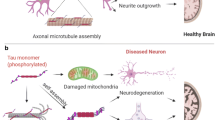
Tauopathy has been long debated in the pathogenesis of AD. Studies based on biomarker analysis show tau protein is more closely linked to AD progression than Aβ. Tau protein is an axonal protein that regulates microtubule stability (Johnson and Hartigan 1999). Normally, tau protein when phosphorylated interacts with and stabilises the microtubules, therefore also known as microtubule-associated protein (MAP). During AD, Tau protein undergoes certain biochemical changes becoming hyperphosphorylated. Hyperphosphorylated form of tau protein dissociates from the microtubules and then aggregates to form neurofibrillary tangles (NFTs) inside nerve cell bodies (Mudher and Lovestone 2002). NFTs disintegrate microtubules within neuronal cells thus destroying their cytoskeleton and ultimately damaging neuronal transport system (Iqbal et al. 2005). Tau protein contains at least forty-five phosphorylation sites, majority of them lie in the C-terminal tail region (residues 368-441) and proline-rich region (residues 172-251) (Hanger et al. 2009). Phosphorylation at C-terminal of tau protein plays more important role so far as AD is concerned. Phosphorylation at Ser262 selectively disrupts binding of tau to microtubules (Fischer et al. 2009), while phosphorylation at Ser202 promotes tau aggregation to form NFTs (Rankin et al. 2005). It seems that phosphorylation at multiple sites of tau protein rather than at single site plays a critical role in the pathology of Alzheimer’s disease (Alonso et al. 2001). Abnormal phosphorylation of tau protein is also implicated in increasing the activity of certain kinases notably glycogen synthase kinase 3-beta (GSK-3β), mitogen-activated protein kinase (MAPK) p38, stress-activated protein kinases, cyclin-dependent kinase-5, p42/44 MAP kinase and mitotic protein kinases; while decreasing the activity of some other enzymes like protein phosphatases (PPs) such as PP1, PP2a, PP2b and PP5 (Mondragón-Rodríguez et al. 2012). Protein phosphatases PP1, PP2A, PP2B and PP5 have been found to de-phosphorylate tau protein at Ser199, Ser202, Thr205, Thr212, Ser214, Ser235, Ser262, Ser396, Ser404 and Ser409. Among all the mentioned PPs, PP2A is considered to be the strongest protein phosphatase (Liu et al. 2005). These entire phenomenons initially hamper the biochemical communication between neurons and later cause neuronal death. Therefore, research should focus on developing drugs with ability to block the abnormal phosphorylation of tau protein which in turn could prevent microtubular destruction as well as neuronal death.
A recent study by Koren and co-workers showed tau protein interacts with ribosomes and aids translation between global transcripts and 5′ Terminal oligopyrimidine (5′TOP) mRNA sequences. But in diseased condition, abnormal tau proteins diminish the effectiveness of translation and foil phosphorylation of ribosomal protein S6 (rpS6) at S240/S244. This reduced activity of rpS6 in turn impairs global translation since the translation of 5′TOP mRNAs coding for ribosomal and translational machinery is affected/reduced (Koren et al. 2019). Thus, there may be some definite link between tau pathology and ribosomal dysfunction in AD which needs to be investigated in future studies.
In addition, studies targeting tau-ribosomes interaction are warranted in exploring effective treatment regimens. Besides phosphorylation, tau protein may also undergo other modifications such as lysine ubiquitylation, lysine acetylation, O-linked N-acetylglucosamine (O-GlcNAc) modification, lysine dimethylation and arginine monomethylation (Du et al. 2018). Nevertheless under pathological conditions, hyperphosphorylation renders tau protein more prone to aggregation thus reducing its affinity to neuronal microtubules and affecting the plasticity of neurons.
Recent research also highlights the role of Apolipoprotein E (apoE) in the pathophysiology of Alzheimer’s disease (Liraz et al. 2013). The ApoE protein encoded by apoE gene is produced by astrocytes in the brain. It is an important protein that transports lipids. ApoE has three alleles namely apoE2, apoE3 and apoE4. ApoE protein is composed of 299 amino acids and its isoforms differ only at amino acid location 112 and 158, which may be arginine or cysteine (Liu et al. 2013). This minor difference is enough to alter the tertiary structure of ApoE protein which disrupts its ability to interact with lipids, Aβ and receptors. Among all the known isoforms of ApoE protein, ApoE4, a hypolipidated version of the protein has been linked to an increased risk of developing Alzheimer’s. ApoE4 allele shows higher percentage in families with sporadic and late-onset Alzheimer’s. Patients with elevated levels of ApoE4 reportedly have an elevated level of accumulated Aβ, hyperphosphorylated tau, reduced neural plasticity and neuropathology (Liraz et al. 2013). Since there occurs accumulation of Aβ in apoE4-positive patients, this led researchers to believe that the two cascades interact with each other and the cross talk between them causes the pathological effects. Occurrence of apoE4 allele negatively hampers the interaction of ApoE4 to lipids thus leading to the proposal that the disease caused due to lipid-related mechanisms (Liraz et al. 2013). ApoE4 not only causes the formation of senile plaques, but also obstructs the body’s means to eliminate it. Normally, the Amyloid plaques and NFTs are removed from the brain through Autophagy (Simonovitch et al. 2016). Under normal conditions, astrocytes of central nervous system (CNS) produce apoE, which protect the brain from harmful protein buildup. Recent research shows that the presence of the apoE4 allele disrupts body’s mechanisms of naturally clearing protein build up through the process of autophagy (Rubenstein 2017). Researchers need to develop drugs with ability to reduce the levels of apoE4 that may allow normal autophagy and prevent abnormal accumulation of proteins in neurons, hence reduce neurotoxicity.
Environmental factors are also implicated in the pathogenesis of various neurodegenerative diseases including Alzheimer’s disease. Many natives of the Island of Guam known as Chamorro suffer from a debilitating neurodegenerative disease with symptoms similar to multitude of neurodegenerative diseases including Alzheimer’s disease (Cox et al. 2016). Persons with this mystery disease showed the presence of Amyloid-β plaques and neurofibrillary tangles in their brains. The non-native people who adopted Chamorro lifestyle also got affected with this disease. The cause of this disease is believed to be an environmental toxin. Initially, there was difficulty in identifying the nature of this neurotoxin because it took years for symptoms to appear after the exposure. Finally, researchers achieved success in isolating this toxin from the seeds of cycad and it was named β-N-methylamino-l-alanine (BMAA). Chamorro people used cycad seeds for making flour, a staple food for them. During 1980s, this toxin was given to macaques which developed acute neurological symptoms. Later in 1990s, Paul Alan Cox, an ethnobotanist proposed that BMAA causes ALS/PDC seen in the natives (Holtcamp 2012). According to him, BMAA is synthesised by cyanobacteria that live in symbiotic relation with the roots of cycad. BMAA gets accumulated in the gametophytes of the cycad seeds, that when ground to flour is consumed by Chamorro people. Later cox showed that BMAA also reaches Chamorro people via animals such as feral deer, flying foxes and pigs who feed on cycad seeds. BMAA undergoes ten thousand times biomagnifications in these animals and thus gets accumulated in the food chain that results in neurological symptoms in Chamorro people. Cyanobacteria that produce BMAA live in many marine ecosystems thus its biomagnifications take place in many marine dwellers like sharks, shellfish and bottom-dwelling fish. The people who consume these animals get BMAA accumulation in their brain and hence develop neurological problems. BMAA can easily cross the blood–brain barrier and reach brain. Cellular mechanism mistakenly takes BMAA for l-serine and incorporates it into proteins thus leading to misfolding, aggregation and apoptotic cell death (Cox et al. 2016). Normally, proteins have an interior hydrophobic part and an exterior hydrophobic part but incorporation of BMAA leads to conformation changes making the exterior part hydrophobic and the interior structure hydrophilic. These external hydrophobic regions of proteins are highly sticky and therefore clump together to form small neurodegenerative protein aggregates. These small aggregates oligomerise to form larger ones that impair cellular functions (Holtcamp 2012). BMAA also promotes phosphorylation of tau protein by causing activation of mGluR5, a metabotropic glutamate receptor, which reduces the activity of protein phosphatase 2A (PP2A). There is a significant decrease of PP2A in Chamorro ALS/PDC brains that increase phosphorylated tau (Cox et al. 2016). Research shows that dietary exposure to BMAA causes deposition of Aβ and formation of neurofibrillary tangles thus acting as an initiator for sporadic Alzheimer’s disease. There is possibility to use l-serine as a therapeutic tool for treatment of AD since it may reduce formation of plaques and NFTs in brain tissue.
Overall, AD seems to be a syndrome rather than a disease since its pathophysiology involves multiple possible models and thus there is requirement of a multifaceted therapeutic approach striking multiple targets in AD for its better clinical and therapeutic outcome. This can be achieved by focusing on ‘magic shotgun drugs” rather than ‘magic bullet drugs”.
Targets for Alzheimer’s Disease
Amyloid Beta
Aggregation of soluble monomeric form of Aβ to form different states of soluble aggregates like dimers, oligomers and polymers is looked as an important factor that causes synaptic loss and cognitive impairment (Lue et al. 1999). Clearing off these toxic aggregates is an imperative therapeutic approach; however, there are certain challenges for instance developing drugs to disintegrate Aβ aggregates can lead to the accumulation of Aβ monomers in the brain thereby causing neurotoxicity; also the strategy to block the formation of Aβ aggregates can lead to the accumulation of monomeric structures that will again trigger neurotoxicity. Nowadays, the best alternative to deal this concern is the development of immunity against Aβ plaques that will completely clear them from brain. Working on same lines, Aβ immunisation performed in mouse model efficiently cleared Aβ plaques from brain but could not improve cognition (Rinne et al. 2010; Wang et al. 2011). In another study, patients suffering from mild-to-moderate AD when vaccinated with Aβ during phase II of clinical trial underwent inflammatory response in CNS (Kuzuhara 2010). However, this inflammatory response has not been fully proven to be responsible for the failure of the anti-Aβ therapy. Janssen and Pfizer’s bapineuzumab were the first attempts to obtain humanised monoclonal antibodies (mAB) with capability to clear-off the amyloid beta from the brain. This drug was tried for thousands of patients with mild-to-moderate AD (Gravitz 2011) but failed to prove its therapeutic potential in phase III of clinical trial. It could not improve the cognition, functional ability and clinical outcome in patients with AD. Research is still going on to find effective antibody against alzheimer’s and in this regard a total of five different anti-Aβ antibodies namely crenezumab, gantenerumab and aducanumab, solanezumab and BAN2401 are being currently tried in preclinical stage of familial AD and asymptomatic individuals at high risk of AD (Salloway et al. 2014; Hung and Fu 2017; Francesco et al. 2019). Gantenerumab, an entirely human recombinant IgG1 monoclonal antibody interacts both with the middle regions as well as the amino-terminal of Aβ. As compared to Aβ monomers, Gantenerumab exhibits higher affinities for Aβ oligomers and Aβ fibrils (Bohrmann et al. 2012). This point is supported by the fact that gantenerumab does not change the plasma Aβ levels in vivo. Intravenous administration of gantenerumab was found to decrease brain Aβ plaques (Jacobsen et al. 2014). However, the cognitive effects of gantenerumab still need to be investigated. Currently, the drug is being investigated in phase II/III trial. Aducanumab, a recombinant human IgG1 monoclonal antibody targets the amino- terminal residues 3–7 of Aβ (Arndt et al. 2018). Aducanumab shows 10,000-fold greater selectivity for soluble Aβ aggregates and insoluble fibrils than for Aβ monomers. Administration of the drug decreased Aβ plaque size in a dose-dependent fashion in young (9-month old) Tg2576 mice but not in aged (22-month old) (Kastanenka et al. 2016). Presently, there are no reports on the cognitive or behavioural effects of Aducanumab. Crenezumab, a humanised anti-Aβ monoclonal IgG4 antibody interacts with multiple species of Aβ, especially fibrillary 16mer assemblies of aggregated Aβ and pentameric oligomeric Aβ (Adolfsson et al. 2012; Zhao et al. 2017). This drug promotes disaggregation and blocks aggregation of Aβ (Ultsch et al. 2016). When injected in Tg2576 mouse model of AD, the drug exhibited no notable inflammatory changes (Fuller et al. 2015). There is deficit of studies pertaining to the cognitive effects of crenezumab in animal models of AD. Currently, the drug is being tested in a double-blind, placebo-controlled clinical trial which may be completed in March 2022 (Tariot et al. 2018). Solanezumab, a humanised IgG1 monoclonal antibody recognises the central region of Aβ (Aβ13–28). Studies carried out in mice and human models indirectly indicate that solanezumab targets the soluble monomeric Aβ because it caused a considerable increase in the levels of total plasma Aβ. Furthermore, studies carried in human brain tissue suggest that solanezumab recognises to Aβ plaques (Bouter et al. 2015). The mouse version of solanezumab known as m266 when injected into the PDAPP transgenic mouse model of AD reversed the memory deficits without affecting brain amyloid plaques (Dodart et al. 2002). There is a possibility that the antibody could target the soluble pool of Aβ (DeMattos et al. 2001). The drug might come out as a potent anti-Aβ regimen in future. BAN2401, another monoclonal antibody specifically binds to soluble Aβ protofibrils (Logovinsky et al. 2016). The antibody showed promising results in an 18-month adaptive phase IIb study carried out in 856 persons with mild or prodromal form of AD. When administered at a higher dose of 10 mg twice a week, the drug caused a significant reduction of brain Aβ accumulation compared to placebo (Osswald 2018) thus indicating the drug could prove effective therapeutic regimen in future.
In addition to the above-mentioned antibody regimens, active anti-Aβ vaccine (antigen) namely CAD106 has also been developed. The vaccine consists of many copies of Aβ1–6 fragment attached to a virus-like particle (an adjuvant carrier). Administration of CAD106 decreased brain amyloid accumulation in two APP transgenic mouse lines without any evidence of inflammatory reactions or increased microhemorrhages (Osswald 2018). In cell cultures, antibodies generated in response to CAD106 administration interacted with Aβ monomers and oligomers and blocked amyloid toxicity (Winblad et al. 2012). Still there is no information available on cognitive effects of CAD106 in animal models of AD. At present, CAD106 is in phase III clinical trials and may come with promising outcome in the near future.
Tau Protein
There are various approaches to target tau protein toxicity notably (I) manipulating phosphatases and kinases, (II) tau vaccination, (III) stabilising microtubules and (IV) blocking tau aggregation. Earlier tau aggregation blocker namely TRx0237 was tested but it failed in phase III clinical trials (Gauthier et al. 2016). Currently, inhibitors like methylene blue and curcumin are being tested. Methylene blue is one of the promising Tau polymerization inhibitors that block the in vitro tau aggregation by trapping monomeric tau in an aggregation-incompetent conformation (Wischik et al. 1996; Panza et al. 2016). In transgenic mouse models of tauopathy, this drug effectively reduced tau pathology and also enhanced cognition (Panza et al. 2016). Methylene blue passes through blood–brain barrier and has been used long in humans; hence, it could serve a promising therapeutic candidate in future. Curcumin, a plant-derived compound isolated from Curcuma longa is another tau polymerization inhibitor that interacts with proteins in β-sheet conformation and block aggregation. The compound has been found to decrease tau as well as Aβ pathology and improve cognitive impairment in animal models (Hamaguchi et al. 2010; Hu et al. 2015). Currently, curcumin is being tested in an 18-month-long phase II study (https://clinicaltrials.gov/show/NCT01811381). In addition, another clinical trial based on combined study of curcumin and exercise is also being recruited (Fitzgerald et al. 2012). Both these trials will assess patients for biomarkers for AD biomarkers as well as for changes on MRI and PET scans. Many other drugs targeting tau aggregation without interfering the stabilisation of microtubules have also been introduced notably emodin, daunorubicin, adriamycin, PHF016 and PHF005 (Pickhardt et al. 2005). Targeting kinases and phosphatases is also looked as a potential candidate for treating neurodegenerative disorders especially AD (Gong and Iqbal 2008). Recent studies have shown sodium-selenate as a possible drug that could treat neurodegenerative disorders by stabilising PP2A in transgenic AD models (Van-Eersel et al. 2010). Moreover, neuroleptics such as trifluoperazine, clozapine and chlorpromazine have also been developed to treat AD (Gong et al. 1996). Among microtubule stabilising drugs, Abeotaxane (TPI 287) is currently being studied in two clinical trials (https://clinicaltrials.gov/show/NCT01966666; https://clinicaltrials.gov/show/NCT02133846). Abeotaxane has shown the ability to pass through blood–brain barrier in mouse model (Fitzgerald et al. 2012). So far as vaccine therapy is concerned, IVIG, the only passive vaccine (Intravenous immunoglobulin) was tried in patients with mild-to-moderate AD but failed in phase III trials (Du et al. 2018). At present, two passive vaccines namely RG6100 and ABBv-8E12; and two active vaccines namely ACI35 and AADvac-1 are being tried in phase I and II clinical trials.
Currently, the preponderance of tau-targeting therapies being tested in trials includes immunotherapies, which have shown promising results in several preclinical trials and may prove effective therapeutics in the near future (https://clinicaltrials.gov/show/NCT01811381). C2 N-8E12 is an antibody that targets amino acids of the tau protein. The drug effectively blocks the pathological tau seeding triggered by exogenous tau aggregates when infused into the brain of transgenic mouse model of tauopathy. C2 N-8E12 diminished the hyperphosphorylated tau and aggregated and also improved cognition (Kfoury et al. 2012; Erin and Congdon 2018). Analogous results were observed when the drug was incorporated systemically. Studies showed no evidence of any adverse immune reaction and furthermore drug administration resulted in the reduction of microglial activation (Yanamandra et al. 2013, 2015). The drug showed promising results in the first phase of clinical trial and is currently being tested in second phase (Congdon et al. 2014; West 2016; Barthélemy et al. 2016). RO 7105705, an antibody most likely recognises pSer409 on the tau protein (Giacobini and Gold 2013). The antibody has not been disclosed so far and is currently being tested in phase I clinical trial. LY3303560 is an anti-tau antibody that possibly recognises a conformational epitope, although this information has not been disclosed so far. Eli lilly has started two phase I clinical trials to test the pharmacokinetics and safety of LY3303560. One trial will be carried in healthy persons and patients with AD, and other trial in patients with MCI or AD (Erin and Congdon 2018). These two trials are expected to reach completion in few years. In the recent times, two new antibodies namely JNJ-63733657 and UCB0107 are also entering the clinical trials. JNJ-63733657 possibly targets the middle region of tau, while UCB0107 this seemingly targets amino acids 235–246 in the proline-rich region of tau (Rogers 2018; https://clinicaltrials.gov/ct2/show/NCT03375697). The field of tau immunotherapy is progressing rapidly and many new trials may come into lime light in the near future.
Alpha Secretases
As far as the role of alpha secretase is concerned it is not involved in the generation of amyloid beta. In fact, α-secretases participate in non-amyloidogenic pathway (Fig. 5) to cleave APP within the amyloid sequences generating a soluble APP ectodomain (sAPPα) through the process termed as “ectodomain shedding”. Function of P3 fragment generated due to the action of gamma secretase in non-amyloidogenic pathway is not clear. Soluble amyloid precursor protein-alpha (sAPPα) is believed to be neuroprotective in the perspective of AD since it hinders amyloid beta peptides formation (Furukawa et al. 1996). sAPPα prevents cultured neurons against the oxygen–glucose deprivation as well as excitotoxicity by blocking calcium currents, enhancing potassium currents and thus stabilising the resting membrane potential. α-secretase activity is mediated by one or more enzymes from the family of disintegrin and metalloproteinase domain proteins (ADAM) with ADAM 9, ADAM 10, ADAM 17 and ADAM 19 being the most probable candidates (Chow et al. 2010). Moreover, alpha secretase causes proliferation of embryonic neural stem cells and promotes synaptic density and improves memory retention (Vella and Cappai 2012). Drugs that enhance alpha secretase activity are considered as promising therapeutics to tame AD. Various drugs acting as indirect α-secretase activators are being tried in different phases of clinical trials to find a treatment for AD. For instance etazolate (EHT 0202), the GABA receptor modulator previously proven to stimulate sAPPα production and protect against Aβ induced toxicity in rat cortical neurons has reached the Phase II of human clinical trials (Marcade et al. 2008; Vellas et al. 2011). PRX-03140, the 5-HT4 agonist proven to stimulate the activity of α-secretase although showed positive results in phase II of clinical trials in 2008 but was not examined further (Sabbagh 2009). Epigallocatechin-gallate (EGCG), a polyphenolic compound obtained from green tea has also proven to act as prominent stimulator of α-secretase through the protein kinase-C (PKC) pathway besides reducing cerebral amyloid deposition in the brain of AD mice. Currently, the efficacy of this drug is being evaluated in phase II/III clinical trial investigations (MacLeod et al. 2015). Recent reports from “Nature Medicine” show that the activity of necrosis factor-converting enzyme (TACE), the stimulator of α-secretase is decreased both in AD patients as well as on the surface of the neurons isolated from mouse model of AD. In addition they also observed an increased activity of 3-phosphoinositide-dependent protein kinase 1 (PDK1) enzyme in the brains of AD patients. It is believed that the later prompts the internalisation of TACE and thus disrupts TACE-mediated α-secretase activity. By blocking the enzyme PDK1 in mouse models using BX912 (PDK1 inhibitor), researchers observed a considerable decrease in the formation of amyloid plaques with an elevation in the levels of sAPPα (Pietri et al. 2013). These results indicate that inhibition of PDK1 enzyme could serve a promising therapeutic approach in future and therefore efforts to develop PDK1 inhibitors may serve a promising therapeutic regimen.

Non-amyloidogenic pathway of Alzheimer’s disease: Alpha secretase cleaves APP to generate the neuroprotective fragment SAPPα towards N terminal via ectodomain shedding. The other remaining fragment of APP fragment is cleaved by gamma secretase into two subfragments P3 and amino-terminal APP intracellular domain (AICD)
Beta Secretase
Beta secretase enzyme playing an important role in Aβ formation is also looked as a possible target to tame neurological disorders but is associated with serious side effects. A study analysing the effect of BACE1 knockout in mice demonstrated significantly higher mortality rate in mice in their 1 weeks of life (Dominguez et al. 2005). Thus, strategy to block this enzyme could prove fatal. Moreover, this enzyme has many important/alternative substrates including the proteins involved in sodium homeostasis and myelination (Wong et al. 2005; Hu et al. 2006). These studies confirm a higher risk of morbidity with the use of BACE1 inhibitors. Now, there arises a question whether the positive modulatory effects of BACE1 inhibitors outweigh its ill effects in the elderly aged people or not. A deep discussion among scientists has lead to an interesting assumption that the partial inhibition rather than complete inhibition of BACE1 could prove beneficial in reducing the levels of Aβ. Various BACE1 inhibitors have been tried in clinical trials but had to be abandoned due to their toxicity. For instance, Eli Lily’s LY2886721 exhibited an initial decrease in the levels of Aβ in plasma but showed liver toxicity due to which its clinical trial was stopped immediately after phase II. Similarly, Eisai’s E2609 demonstrated a considerable reduction in plasma Aβ levels during Phase I of clinical trial but its Phase II trials have not been tried yet (Wolfe 2012; Folch et al. 2018). In an another attempt, the development of HPP854 by TransTech Pharma and RG7129 by Roche had to be stopped immediately after phase I clinical trial due to some undisclosed reasons. The ongoing clinical trial sponsored by Merck Sharp and Dohme Corp evaluates the efficacy of Verubecestat in patients with prodromal Alzheimer’s disease. This clinical trial (NCT01953601) is expected to generate its outcome in February 2019. In addition, AstraZeneca and Eli Lilly and Company have joined their hands together for co-developing lanabecestat (AZD3293). The crucial Phase II/III clinical trial of lanabecestat started in late 2014 and is intended to recruit 1500 patients and will probably end in May 2019 (https://www.reuters.com/article/2014/12/01/health-alzheimers-astrazeneca-eli-lilly-idUSL6N0TL0ST20141201).
Gamma Secretase
Gamma secretase, although a much anticipated target for treating AD, has been shown to cleave a wide range of substrates. The most prominent substrates for this crucial enzyme include Notch, an important cell surface receptor pivotal for cellular development and differentiation, whose role is also implicated in tumour suppression (Sorensen and Conner 2010). Inhibition of γ-secretase has been found associated with potential toxic effects, for example, Eli Lily’s semagacestat (LY450139) was abandoned after phase III due to its failure to stop disease progression and worsening the cognition (Doody et al. 2013). Studies also warn about the side effects like skin cancer due to γ-secretase inhibition, most probably due to concomitant inhibition of Notch signalling. Bristol-Myers-Squibb’s Avagacestat (BMS708163) was abandoned after Phase II trials due to side effects like worsening of cognition and the risk of skin cancer (Coric et al. 2012; Golde et al. 2013). Here it is worth mentioning that even the Notch-sparing inhibitor ELND006 developed by Elan Corporation when subjected to clinical trial was found to cause liver toxicity and was abandoned (Hopkins 2011). Another drug, Tarenflurbil used as γ-secretase modulator although showed promising outcome during phase II clinical trial, was discontinued due to poor response in third phase of clinical trial (Green et al. 2009). Similarly, CHF-5074 (γ-secretase modulator) developed by Chiesi successfully reached phase II of clinical trial (NCT01303744) but was abandoned due to some hidden reasons. The futuristic strategy of γ-secretase inhibition will largely depend on the development of APP-specific γ-secretase inhibitors with property to spare Notch signalling.
Acetylcholinesterase
Acetylcholinesterase (AChE) belongs to carboxylesterase family of enzymes. It is the primary cholinesterase in the body that catalyses the breakdown of neurotransmitters such as acetylcholine and other choline esters. Acetylcholinesterase is a key enzyme in the cholinergic nervous system synthesised in the muscle cell and then released into the neuromuscular junction/synaptic cleft where it functions as the terminator of synaptic transmission. AChE cleaves acetylcholine molecules into acetyle and choline. Later, the choline re-enter the neuronal cell where it is re-used to synthesise acetylcholine (Fig. 6). During the pathology of AD, there is deterioration of many cholinergic neurons in the brain which results in the profound decline of acetylcholine (Du et al. 2018). Thus, AD patients produce less acetylcholine (compared to normal person), which hampers the normal neurotransmission and thus producing clinical manifestations of AD like memory loss. The strategy to inhibit the function of acetylcholinesterase using acetylcholinesterase inhibitors is looked as a promising approach to raise the level of acetylcholine and promote continued stimulation of the muscles and glands that potentiate the parasympathetic activities such as constriction of pupils of the eyes, vasodilatation, increased production of saliva, sweat, and tears, slow heart slow heart rate, constriction of bronchioles and mucus secretion in the respiratory tract (Samii et al. 2004). Drugs like donepezil, galantamine and rivastigmine are commonly used as AChE inhibitors for providing symptomatic relief (Jacobsen et al. 2005; Mehta et al. 2012). There has been a continued search for obtaining efficient inhibitors of acetylcholinesterase and thus researchers have explored many new/modified synthetic and natural compounds/drugs with potential anti-acetylcholinesterase activity. The presence of peripheral anionic site (PAS), in addition to the catalytic site (CS) on acetylcholinesterase, has been implicated in promoting formation of amyloid fibril and its co-localization. Novel flavonoid derivates capable of binding to both the PAS and CS site of acetylcholinesterase have been designed and can inhibit acetylcholinesterase much better than conventional rivastigmine and donepezil (Sheng et al. 2009). Khan and co-workers used chemical as well as computation-based approaches to evaluate the derivatives of four flavonoids namely quercetin, rutin, kaempferol and macluraxanthone. They found macluraxanthone and quercetin derivatives as very good inhibitors of acetylcholine esterase (Khan et al. 2009). Various modified novel carbamates tested in silico and in vitro have been found to possess prominent AChE inhibitory activity (Roy et al. 2012). In addition, pyridopyrimidine, a novel natural compound has shown greater AChE inhibitory action than the conventional drug galantamine (Kumar et al. 2016). The hybrids of donepezil and aminopyridine namely pyridonepezil and 6-chloro-pyridonepezil, respectively, have been found to be more potent inhibitors of AchE than the single donepezil molecule (Varadaraju et al. 2013). These molecules are considered as the dual inhibitors of AchE that bind to both the PAS and CS site of acetylcholinesterase. Developing such therapeutics with better efficacy and safety could serve as a promising strategy in ameliorating AD.

Prolonged activation of acetylcholine esterase caused decline of acetylcholine (neurotransmitter). The strategy to inhibit acetylcholinesterase using acetylcholinesterase inhibitors is looked as a promising approach to raise the level of acetylcholine and promote continued stimulation of the muscles and glands
N-methyl-D-aspartate Receptor
N-methyl-D-aspartate receptor also called as the NMDA receptor or NMDAR is an ion channel protein and a glutamate receptor present in the membrane of nerve cells. NMDAR is activated when glutamate and/or glycine (or d-serine) binds to it; and in its activated form NMDAR permits the flow of positively charged ions through the cell membrane of nerve cells. This receptor is very important for controlling synaptic plasticity as well as the memory function. During Alzheimer’s disease, there is hyperactivation of NMDAR due to glutamate binding which leads to excessive influx of Ca2+ ions through it (Lipton 2005). Here it is worth mentioning that glutamate acts as the most important excitatory neurotransmitter in the brain and glutamate-mediated synaptic transmission is vital for the normal functioning of the nervous system (Dingledine et al. 1999). Calcium influx triggers hyperactivation of enzymes including the phospholipases, cyclooxygenase-2 (COX-2), lipoxygenases (LOX), proteases and nitric oxide synthase (NOS) (Tan et al. 2009a, b; Ezza and Khadrawy 2014). Glutamate-mediated overactivation of NMDAR and subsequent Ca2+ influx is associated with mitochondrial dysfunction, free radical generation and neurodegeneration (Fig. 7).

Glutamate induced activation of NMDAR, calcium influx, free radical generation and subsequent neuronal death
Activated phospholipases cause hydrolysis of essential phospholipids from neuronal membrane and subsequent accumulation of free fatty acids (FFAs), diacylglycerol (DAG), platelet activating factor (PAF’s) and lysophospholipids. FFAs, DAG and lysophospholipids have detergent-like effect on normal membranes and can uncouple oxidative phosphorylation thereby producing changes in membrane permeability. PAF is pro-aggregatory and may cause adhesion and activation of leukocytes and hence produce an inflammatory reaction at endothelial cell surface. FFAs will trigger arachidonate pathway and generate inflammatory mediators including prostaglandins, leukotrienes, leukotrienes and thromboxanes. Prolonged activation of arachidonate pathway triggers increased production of free radicals, lipid peroxidation and hence oxidative damage to membrane-bound proteins. Phospholipase mediated phospholipid degradation also sets stage for calcium influx and PKC activation, which has deeper implications in neurodegeneration (Asaoka et al. 1992).
Activated lipoxygenases (LOXs) catalyse oxidation of arachidonic (AA) to various bioactive lipids implicated in neurodegeneration. Briefly, Lipoxygenases (LOXs) oxidise AA to hydroperoxyl derivatives including 5-hydroperoxyeicosatetraenoic acids (HPETEs). These derivatives upon reduction form corresponding hydroxyeicosatetraenoic acids (HETEs) and leukotriene (LT) via 5-lipoxygenase, lipoxins and hepoxilins. LOXs peroxidize membrane lipids and lead to structural changes in the cell. 15-LOX-1 is the major enzyme which is responsible for membrane lipid peroxidation (Brash 1999). 5-HPETE inhibits synaptosomal membrane Na+, K?-ATPase activity (Foley 1997).
Activated COX-2 oxidises dopamine to a highly reactive form dopamine-quinone which can react with cysteinyl residues of target proteins, and transform them. Transformation could in turn alter the function of proteins leading to cell death. COX2 also increases production of prostaglandin E2, which in turn leads to increased production of reactive oxygen species (Bazan 1999).
Activated protease converts xanthine dehydrogenase to xanthine oxidase. The latter is a key enzyme in the production of free radicals such as hydroxyl and superoxide. The brain is highly sensitive to free radicals since it lacks normal free radical scavengers. Free radical acts on the phospholipid of neuronal membrane and destroys them. Ca2+ also activates NOS which subsequently increases nitric oxide production. The latter is a gaseous free radical and mediates the excitotoxicity and neurodegeneration (Gagliardi 2000).
Excess of Ca2+ influx due to NMDAR overactivation also triggers opening of mitochondrial permeability transition pore (PTP) through which cytochrome c (Cyt c) and apoptosis-inducing factor (AIF) comes out into cytoplasm and stimulates apoptotic pathway leading to programmed cell death (Hibiki and Giovanni 2017). Prolonged calcium influx through continuous opening of NMDA receptors disrupts mitochondrial membrane potential, disrupts the oxidative phosphorylation thus reducing ATP production and making cell more susceptible to death insults (Fiskum et al. 2003). Due to disruption of energy metabolism, the energetically compromised neurons become depolarized and cannot maintain ionic homeostasis (Zhou and Danbolt 2014).
Drugs/molecules which act as antagonists of NMDA receptor are therapeutically considered beneficial in many neurological disorders like dementia, stroke and neuropathic pain syndromes (Gitto et al. 2014). At present, memantine is the only drug marketed as NMDA receptor antagonist. A wide range of molecular docking studies are being carried out to obtain novel/active ligands against this receptor in AD. Examples of some prominent ligands confirmed by docking experiments include triazolylamidine derivatives (Espinoza-Moraga et al. 2012); 3-substituted-1H-indoles (Colotta et al. 2012); phenyl-amidine; 1-benzyl-1,2,3,4-tetrahydro-b-carboline (Parson et al. 2013); and 3-hydroxy-1H-quinazoline-2, 4-dione derivatives (Abreu et al. 2013). The amino acid glycine binds to NR1 subunit of NMDA receptor and has been identified as a co-agonist of NMDA. Studies are being conducted to find out the molecules that could block the glycine binding NR1 subunit of NMDA receptor (Krueger et al. 2009). In addition, studies based on molecular docking have revealed that ifenprodil and similar compounds posses strong activity in blocking NR2B unit of NMDA (Gitto et al. 2008).
Muscarinic ACh Receptors
Muscarinic acetylcholine receptors (mAChR) form one of the G-protein-receptor complexes in the cell membranes of certain neurons and other cells and act as ACh receptors at various locations including the central nervous system (CNS). They are stimulated by neurotransmitter Ach, released from postganglionic fibres in the parasympathetic nervous system (PNS). mAChR are of five types represented as M1-M5 and are implicated in motor control and learning process. The M1-type mAChR present in the cerebral cortex and hippocampus play important role in memory, cognitive processing and learning which become impaired in Alzheimer’s disease (Caulfield and Bridsall 1998). These cholinergic deficits observed in AD can be restored via cholinergic activation, which can be done by the use of muscarinic agonists. Over many years, M1 subtype agonists/drugs such as AF102B, AF150, AF267B and AF292 have been tried in AD patients. Among these compounds AF267B has been found to have excellent pharmacokinetic profile and can also cross the blood–brain barrier, whereas the compounds namely AF150(S), AF102B and AF267B are associated with neurotrophic effects, elevated non-amyloidogenic APP and decreased Aβ (Davie et al. 2013). In Alzheimer’s disease, formation of amyloid beta reduces the potential of mAChR receptors to transmit signals, thereby leading to diminished cholinergic activity. Reports suggest that the strategy to activate M1 mAChRs could ameliorate AD pathology besides restoring cognitive functions and decreasing hyperphosphorylated form of tau protein (Foster et al. 2014). Here, it is worth mentioning that even though a few M1 agonists improved cognition in the initial phase, but failed after reaching clinical trial due to their non-specific nature (Melancon et al. 2013). 77-LH-28-1, an M1 allosteric candidate from GlaxoSmithKline (Harlow, UK) has shown great CNS penetration and promising pharmacological profile. In addition, two other M1 selective agonists namely VU0364572 and VU0357017 developed by “Vanderbilt Centre for Neuroscience Drug Discovery”, (Nashville, TN, USA) have been tested on various animal models and cell lines and found effective on many parameters (Kumar et al. 2016). Although still in infancy, the strategy to develop mAChRs activators may serve a good approach in taming AD.
Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder damaging mainly the motor system and affects about 1.5% of the world population over the age of sixty (Lozano et al. 1998). This disease is characterised by the death of 70–80% of dopaminergic neurons in the substantia nigra—the important part of mid-brain, thus resulting in the decreased secretion of a neurotransmitter i.e. “dopamine” in these areas of brain. Since neurons of this region of brain control the voluntary movements therefore there occurs degeneration in four cardinal symptoms namely postural imbalance, bradykinesia, muscular rigidity and resting tremor (Bhat et al. 2015). All together these motor symptoms represent “Parkinsonism” or “Parkinsonian syndrome” (Fig. 8).

Pathophysiology of Parkinsonism and its cardinal signs
Sometimes also called as syndrome, Parkinson’s disease has a very complex pathophysiology and involves not only the dopaminergic loss of neuronal cells but also the loss of noradrenergic, cholinergic and serotonergic neurons. Research shows that the additional loss of noradrenaline (NA) neurons of the locus coeruleus, the chief source of NA in the brain could cause motor and non-motor dysfunctions. Furthermore, the use of selective agonists and antagonists of noradrenaline alpha receptors, scientists have revealed that α2 receptors have an important role in controlling motor activity and that targeting α2 receptor with antagonists could prove pivotal in improving the motor symptoms as well as l-Dopa-induced dyskinesia. Damage or loss of NA neurons in PD influences all PD symptoms and the strategy to add NAergic drugs to dopaminergic medication seems a promising approach in the treatment of the disease.
Serotonin (also known as 5-Hydroxytryptamine), an another neurotransmitter plays important role in developing PD by affecting several motor and non-motor functions thereby causing symptoms like tremor, cognition impairment, depression, psychosis, as well as L-DOPA-induced dyskinesia (Huot et al. 2017). Declined levels of 5-HT were observed in the prefrontal cortex (PFC) up to 18 weeks following an acute injection of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mice (Ansah et al. 2011). In another study, reduction in the levels of 5-HT transporter (SERT) in the cortex and anterior cingulate was reported post unilateral striatal lesions in the macaque monkey (Sanchez et al. 2013). Furthermore, decline of serotonin transporter (SERT) immunoreactive axons in the (prefrontal cortex) PFC declined 5-HT-immunoreactivity in the median raphe neurons, or reduced PFC SERT binding capacity has also been observed in PD brains (Guttman et al. 2007; Haapaniemi et al. 2001). PD patients suffer a loss of about 25% serotonergic receptors (HT1A) at median raphe nucleus, and this is concurrent with the severity of resting tremor (Doder et al. 2003), which indicates that 5-HT projections in mid-brain have more implication in triggering PD tremor than loss of nigrostriatal DA-projections. Experiments in MPTP model of PD suggests that 5-HT turnover in the PFC may have an important role in executive dysfunction (Maiti et al. 2016). Several investigators in PD show a strong relation between decline of 5-HT and depression (Tan et al. 2011); however, further research is needed in elucidating the role of 5-HT with the progression of PD.
A broad band of cell clusters called nucleus basalis of Meynert (nbM) present within the basal forebrain subventricular region is principally cholinergic in nature. Acetylcholine is found downregulated in many neurological diseases, including PD (Tagliavini and Pilleri, 1983, Tan et al. 2011)). In fact, evidences revealing neuronal loss in the nbM region of PD patients indicate a definite involvement of cholinergic system in PD pathophysiology (Tagliavini and Pilleri 1983; Liu et al. 2015). In addition, PD patients with cognitive decline show the presence of lewy bodies as well as neuronal loss in nbM region of brain of suggesting the involvement of cholinergic system in the cognitive loss.
PD involves accumulation of abnormal protein aggregates called lewy bodies inside the neuronal cells of substantia nigra (mid-brain) (Kalia and Lang 2015). Lewy bodies appear as spherical masses that displace other cell components. They are composed of eosinophilic cytoplasmic inclusions forming a dense “core” surrounded by a “halo” of radiating fibrils composed of alpha-synuclein. Parkinson’s disease (PD) is sometimes known as synucleinopathy due to an abnormal accumulation of alpha-synuclein protein in the brain in the form of lewy bodies (Galpern and Lang 2006). In addition to alpha-synuclein, Lewy bodies also contain proteins like ubiquitin, neurofilament protein and alpha-B-crystalline (Engelender 2008). Sometimes tau proteins may also be present in lewy bodies (Arima et al. 1999; Ishizawa et al. 2003). Alpha-synuclein, the major culprit in the pathogenesis of PD seems to get transferred from one neuronal cell to another cell through many pathways such as direct membrane penetration, endocytosis, exosome-mediated transfer, trans-synaptic dissemination and receptor-dependent uptake (Lashuel et al. 2013). An elevated level of alpha-synuclein in the brain neurons of PD patients is the indicative of defective alpha-synuclein clearance which contributes to its pathogenesis. Normally, alpha-synuclein can undergo degradation in neurons by two ways i.e. through ubiquitin–proteasome system (UPS) and the autophagy/lysosomal pathway (ALP) (Dehay et al. 2015). There is presence of a chaperone-mediated autophagy (CMA) recognition motif 95VKKDQ99 (KFERQ like) in the alpha-synuclein protein that allows its interaction with cytosolic chaperone “HSC-70″ and then translocation into the lysosome via lysosome-associated membrane receptor protein, LAMP2a (Cuervo et al. 2004). In vivo studies confirm that the normal soluble form of α-synuclein is mainly degraded by UPS while the complex aggregated form is degraded by ALP (Ebrahimi-Fakhari et al. 2011). Research has shown depletion in proteasome components as well as reduced lysosome number in sporadic PD brains and in both toxic and genetic rodent models (Dehay et al. 2010). There occurs a vicious cycle leading to accumulation of alpha-synuclein in brain cells and the formation of lewy bodies which in turn hinders protein clearance thereby leading to neuronal damage. Currently, there is no cure for Parkinson’s disease. However, some medications like dopamine agonists notably L-DOPA (levodopa) are being used, which becomes less effective after long-term use and produces side effects marked by involuntary writhing movements. With the passage of time there occurs massive neuronal loss. These medications become less effective while at the same time they produce complications marked by involuntary writhing movements (Sveinbjornsdottir 2016). Scientists mostly consider PD a non-genetic disorder; however, around 15% of individuals with PD show first degree relative that has the disease (Samii et al. 2004). Classical treatment strategies for Parkinson’s disease include the use of three types of therapeutic approaches namely dopamine agonists, dopamine antagonist (e.g. inhibitors of monoamine oxidase) and inhibitors of catechol-O-methyltransferase. In 2005, Rascol and co-workers reported potential therapeutic role of MAO-B inhibitor “rasagiline” in PD patients with motor fluctuations. Rasagiline drug was compared against an agent from another class-the catechol-O-methyltransferase inhibitor “entacapone”. Both drugs exhibited comparable benefit (Rascol et al. 2005). In addition, techniques like gene therapy and cell transplantation are being tested in animal models to find an effective treatment for PD. However, such treatment strategies are still in infancy and may take several years to become successful.
Targets for Parkinson’s Disease
Alpha-Synuclein
The first predominantly hereditary mutation described in PD was that of A53T encoding alpha-synuclein (Polymeropoulos et al. 1997). Mutated form of alpha-synuclein protein aggregates inside the dopamine neurons of the substantia nigra and other brain-stem neurons leading to lewy body formation thereby making neurons prone to oxidative stress/cell death. Evidences indicate that toxicity of alpha-synuclein is mediated by Sirtuin-2 protein (an important member of HDAC family) under both the in vitro as well as in vivo transgenic Drosophila model of PD (Outeiro et al. 2007). Being a crucial protein in the pathogenesis of PD, alpha-synuclein is looked as a novel drug target for the possible treatment of this dreadful disease. Currently, one of the most trusted strategies to tame PD is the use of antibodies that target alpha-synuclein protein. Positive results from in vivo mouse model studies have led various pharmaceutical companies like Roche to start clinical trial for PRX002, the monoclonal antibody directed against α-synuclein. The antibody was initially developed by Elan Pharmaceuticals (patent#US7910333). In another example, active immunisation with Affitope PD01 (Affiris, patent O2009103105) in 32 PD patients was found safe in a first pilot study (Dehay et al. 2015). Moreover, passive Immunisation involving the use of a novel monoclonal α-syn antibody (9E4) against the carboxy-terminus of alpha-synuclein was shown to traffic into the central nervous system (CNS) and improves the deficits caused by alpha-synuclein aggregation. Various in vivo experiments showed that 9E4 cross into the CNS and bind to cells that display alpha-synuclein accumulation and promotes alpha-synuclein clearance via the lysosomal pathway (Masliah et al. 2011). In future, drugs preventing alpha-synuclein aggregation may become an effective strategy to combat PD.
Parkin
Ubiquitination of substrates is a highly regulated biochemical mechanism involving three important enzymes namely E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme and E3 ubiquitin ligase 9 (Hershko and Ciechanover 1998). E3 ubiquitin ligases play an essential role in the recognition of substrate and thus contribute to the specificity of ubiquitin reaction. Defective parkin-mediated ubiquitination may disrupt the targeting of specific substrates for degradation (Tan et al. 2009a, b), thereby causing accumulation of toxic proteins in the cell followed by cell death (Fig. 9). Parkin is composed of 465 amino acids and acts as E3 ligase in the ubiquitin–proteasome system (Shimura et al. 2000; Kahle and Haass 2004) and is widely neuroprotective in action. Parkin deficiency may affect cell survival through complex mechanisms. Parkin mediates autophagic degradation of mitochondria during mitochondrial depolarization (Narendra et al. 2008). Autosomal recessive loss-of-function mutations in PARK2 gene cause functional inactivation of parkin thus leading to degeneration of catecholaminergic neurons and a familial form of Parkinson’s disease. There are evidences which indicate that there may be some correlation between the mitochondrial function of parkin and its neuroprotective role. The role of parkin in blocking apoptosis has been widely reported and may involve the basic alterations in the threshold for the release of apoptotic cytochrome c. Parkin blocks the basal and apoptotic stress induced translocation of Bax to the mitochondria. Furthermore, its apoptotic function was retained by an engineered ubiquitination-resistant form of Bax. In addition, bax knocked out cells complemented with lysine-mutant bax did not manifest the antiapoptotic effects of parkin that were observed in cells expressing wild-type bax. Also the parkin-deficient neurons are more sensitive to apoptosis, exhibiting 74% increase in the levels of caspase 3/7 (Johnson et al. 2012). In addition to its role in ubiquitin–proteasome system (UPS), parkin has many other functions for instance it leads to monoubiquitylation of epidermal growth factor receptor pathway substrate 15 (Eps15). This monoubiquitylation event in turn blocks the interaction between Eps15 and ubiquitinated EGF receptor (EGFR) and promotes EGFR internalisation. Impaired function of parkin may disrupt this process leading to enhanced endocytosis and degradation of the EGF receptor thereby decreasing neuronal survival (Fallon et al. 2006). Blocking parkin expression promotes oxidative damage while increasing parkin expression diminishes oxidative damage (Shin-ichiro et al. 2013). Based on these lines, strategy to enhance the non-proteasomal ubiquitinating functions of parkin will contribute significantly to neuronal survival. Gene therapy has been proposed as a promising approach to modulate the effects of aberrant protein aggregation and misfolding for instance virus-mediated delivery of parkin effectively reduces the neuronal toxicity caused due to overexpression of alpha-synuclein and it is supposed that lentiviral delivery of beta-synuclein may also confer similar neuroprotective benefits.

Figure depicting the altered/mutated parkin affects normal ubiquitination process thereby inhibiting the proteasome-mediated breakdown of selected proteins. Accumulation of the latter causes neurotoxicity and hence neurodegeneration
Molecular Chaperones
Molecular chaperones are an important class of proteins that interact with other proteins and help them in acquiring their native/stable conformation (Hart et al. 2011). In neurons, there is an extensive network of chaperones and co-chaperones that mediate protein folding and re-folding. They also interact with protein degradation cascades like ubiquitin–proteasome pathway or autophagy to eliminate the wrongly folded and potential pathogenic proteins. Thus, deregulation of chaperone expression is critical for neurodegeneration. Studies demonstrated the role of chaperones like Hsp70, Hsp90, Hsp40, Hsp60 and Hsp27 as part of lewy bodies in PD (Ebrahimi-Fakhari et al. 2013). Hsp70 is believed to have critical role in maintaining α-synuclein’s folding (Auluck et al. 2002) and also mitigates α-synuclein toxicity. Exogenous overexpression of Hsp70 and other chaperones has critically proven neuroprotective in PD models. GRP78/BiP (an HSP70 located in the endoplasmic reticulum) diminishes α-synuclein-mediated neurotoxicity by downregulating Endoplasmic Reticulum (ER) stress mediators and the level of apoptosis. GRP78/BiP also promotes the survival of nigral tyrosine hydroxylase (TH)-positive cells and elevates striatal DA levels. There is formation of a complex between GRP78/BiP and α-syn that may possibly contribute to prevent neurotoxicity caused due to α-synuclein. Molecular chaperones GRP78/BiP have a definite neuroprotective role in α-synuclein-triggered neurodegeneration (Marina et al. 2012). These data, together with the results from age-related studies, highlight the importance for developing drugs to induce elevation of endogenous GRP78 in order to increase cellular survival and extend functional longevity. BIX (a selective inducer of BiP, BiP Inducer X (BIX) has been shown to selectively induce the GRP78 mRNA and to modulate the ER stress response in cells, thus promoting the survival of neuronal cells undergoing degeneration associated with activation of the UPR. This has further implied that augmentation of GRP78 is a feasible therapeutic approach for the treatment of neurodegeneration (Marina and Oleg 2013). Co-expression of TorsinA (a protein with homology to Hsp104) (McLean et al. 2002a, b), Hsp27 (Zourlidou et al. 2004), Hsp40 (McLean et al. 2002a, b) or Hsp70 (Klucken et al. 2004) remarkably diminishes α-synuclein levels and neurotoxicity. Studies prove that there occurs a transient and reversible interaction between the substrate-binding domain of HSP-70 and the core hydrophobic region of soluble α-synuclein intermediates (Luk et al. 2008). While elucidating the role of small Hsps (Hsp20, Hsp27, HspB8, HspB2B3 and αB-crystallin) with both mutant and wild-type α-synuclein, it was found that all HSPs interacts transiently with different forms of α-synuclein and blocks the formation of mature α-synuclein (Bruinsma et al. 2011). Molecular chaperones might thwart neurotoxicity via different ways like e.g. by facilitating disease protein degradation or sequestration, blocking inappropriate protein interactions or by blocking downstream signalling events responsible for neurodegeneration. Thus, cellular depletion and the subsequent loss of chaperone function may promote neurodegeneration. Targeting chaperones with drugs that increase their expression is regarded as a promising strategy for treating PD (Muchowski and Wacker 2005a, b). Chemical chaperones or the compounds with direct chaperone activity like geldanamycin, trehalose, 4-phenylbutyrate, celastrol and mannosylglycerate are being tested as potential anti-PD therapeutics. For instance, trehalose (a disaccharide) demonstrates direct interaction with client proteins and can also promote autophagy-mediated protein clearance in different models of neurodegenerative diseases. The compounds mannosylglycerate, 4-phenylbutyrate and mannitol can markedly ameliorate α-synuclein aggregation and toxicity in different PD models (Ebrahimi-Fakhari et al. 2013). Chemical chaperones with low toxicity could be the ideal candidates for future drug development to treat PD.
A2A Receptors
In addition to dopamine, many other neurotransmitters such as 5-hydroxytryptamine, norepinephrine, adenosine, glutamate and acetylcholine are implicated in PD. Researchers try to explore the role of non-dopaminergic therapies for PD. Modulation of neurotransmitters that act downstream of dopamine in the striatal outflow pathways is a promising target. A good example of such a strategy includes the inhibition of adenosine 2A (A2A) receptors. Briefly, the A2A receptor belongs to the G-protein-coupled adenosine receptor (GPCR) family and is expressed majorly in striatum (Cristalli et al. 2009; De-Lera-Ruiz et al. 2013). A2A receptors are also expressed at lower levels in other parts of brain including cerebral cortex, hippocampus, motor nerve terminals, nucleus tractus solitarius and glial cells. Activation of A2ARs enhances the release of several neurotransmitters, for instance dopamine, acetylcholine and glutamate but inhibits the release of gamma aminobutyric acid (GABA). A2A receptor activation also modulates neuronal excitability and synaptic plasticity, and affects various behaviours including sleep–wake cycle, locomotor activity, depression, anxiety, learning and memory. At the cellular level, A2ARs are localised predominantly in the soma of GABAergic (enkephalin-containing, dopamine D2 receptor-expressing) striato-pallidal projection neurons and to a lesser extent in asymmetrical excitatory synapses at the dendrites of cortico-striatal terminals. At the molecular level, the A2AR has been shown to interact with other neurotransmitters and neuromodulator receptors (possibly through molecular dimerization) including metabotropic glutamate receptor subtype 5 (mGluR5), adenosine A1 receptor (A1R), dopamine D2 receptor (D2R), cannabinoid receptor type 1 (CB1) and facilitatory nicotinic acetylcholine (Ach) receptor. These interactions expand the range of possibilities used by adenosine to interfere with neuronal function and communication (De-Lera-Ruiz et al. 2013). The strategy of blocking A2A receptors itself has shown a moderate effect on the symptoms of PD but when used in combination with levodopa and dopamine has improved the therapeutic outcome significantly (Bara-Jimenez et al. 2003). Results obtained from animal models also indicate that the use of adenosine receptor blockers could protect neurons in PD (Chen et al. 2001). Till now, clinical research testing A2A antagonists as therapeutic candidates for PD continues to evolve from drugs formerly discontinued (i.e. preladenant and vipadenant) to new derivatives in development (like tozadenant, ST1535, ST4206, PBF-509 and V81444) and the relatively older drug istradefylline, which has been already licensed in Japan as an anti-parkinsonian drug (Pinna 2014).
GFLs
GFLs are the proteins that regulate the development and maintenance of the nervous system. GFLs support and restore multiple neuronal populations such as dopaminergic, hippocampal, sensory, motor, enteric, basal forebrain, sympathetic and parasympathetic neurons. Thus, GFLs have a great potential for curing diseases caused due to neurodegeneration. Till now four GFLs namely GDNF, neurturin (NRTN), artemin (ARTN) and persephin (PSPN) have been characterised. GFLs mediate their effects by a receptor complex which consists of the signal-transducing module receptor tyrosine kinase known as RET and the ligand-binding co-receptor i.e. GFRα. Like in the case of other RTKs, RET has an intracellular tyrosine kinase domain (Gill et al. 2003) and a unique extracellular domain (ECD) composed of a cysteine-rich region and four cadherin-like regions (He et al. 2005; Slevin et al. 2005). RET does not interact with GFLs directly, but GFLs bind with GFRα co-receptors. GDNF preferably binds to GFRα1; ARTN binds to GFRα3; NRTN binds to GFRα2 and PSPN binds to GFRα4. Additionally, GDNF can activate RET through GFRα2, and NRTN through GFRα1 while as ARTN shows weak binding to GFRα1 in vitro (Sidorova and Saarma 2016). GDNF, a distant member of the transforming growth factor-beta (TGFβ) superfamily was discovered in 1993 and since then it has attracted considerable attention for its potential to protect and repair dopamine-containing neurons, which get degenerated in PD. GDNF shows specific binding to GDNF family receptor α1 (GFRα1) and forms a complex which binds to and signals through the transmembrane receptor tyrosine kinase, RET. GFLs like GDNF and NRTN have been tested in several clinical trials in patients with PD. They showed promising results in the early phase of clinical trial but failed in later phase due to poor pharmacokinetic properties like inability to penetrate blood–brain barrier, high affinity for extracellular matrix, etc. Besides, there are some obstacles associated with approaches such as neurosurgery, or use of encapsulated cells, and viral vectors to deliver GFLs into the brain. Even if there are limitations but GFLs especially GDNF is looked as an attractive therapeutic target for the treatment PD. The strategy to develop GFL mimetics with ability to activate GFL receptors is believed to provide good therapeutics to ameliorate Parkinson’s disease (Bespalov and Saarma 2007). Sidorova and co-workers screened 18,400 compounds for developing GDNF mimetics and identified BT13 as a potential compound that selectively targets GFL receptor, RET and activates downstream signalling pathways. Like NGF and ARTN, BT13 selectively promotes neurite outgrowth from the peptidergic class of adult sensory neurons in culture. BT13 effectively reduces mechanical hypersensitivity and also normalises the expression of sensory neuron markers in dorsal root ganglia of rat model with neuropathic pain (Sidorova et al. 2017). Bespalov and co-workers reported the development of another compound namely BT18 that selectively activates GFL receptors, eases pain and repairs the damaged dorsal root ganglion (DRG) neurons in rat models of neuropathic pain (NP) (Bespalov et al. 2016). XIB4035 is a novel molecule thought to be an agonist at the GDNF family receptor α1 (GFRα1). Like GDNF, XIB4035 induces RET autophosphorylation and promotes neurite outgrowth in Neuro-2A cells (Tokugawa et al. 2003). XIB4035 is unable to activate RET in the absence of endogenous ligand, i.e. GFL. Recent studies indicate that it enhances GFRα family receptor signalling in conjunction with ligand stimulation. It has resulted in the effective treatment of small-fibre neuropathy in mouse models of the disease (Hedstrom et al. 2014; Hsieh et al. 2018). GDNF agonists may act as new class of therapeutics for treating PD but this area of research needs more advancement.
LRRK2
Leucine-rich repeat kinase 2 (LRRK2) is a multidomain and multifunctional protein expressed widely. It is also named as dardarin and is encoded by PARK8 gene in humans. Besides having kinase activity, LRRK2 can also take part in intracellular signalling as a scaffold protein by bringing together heterologous complexes via its various protein–protein interacting (PPI) domains. Mutations in LRRK2 gene are the major cause of inherited and sporadic form of Parkinson’s disease. There is substantial overlapping between clinical familial LRRK2-linked PD and idiopathic PD. Exploring LRRK2 function may provide deep insights into both familial as well as idiopathic form of PD (Haugarvoll et al. 2008; Healy et al. 2008). The Gly2019Ser mutation is one of the prominent LRRK2 mutations that lead to Parkinson’s disease. Recently, genome-wide gene expression study was conducted to compare G2019S-associated PD and idiopathic PD. This study identified deregulations in mechanisms associated with complement and coagulation cascades, cell adhesion molecules, extracellular matrix organisation and hematopoietic cell lineage. Although this study generated deep insights into the probable LRRK2-associated mechanisms in Parkinson’s disease, the large difference between the blood transcriptome profiles and G2019S-associated PD and idiopathic PD supports the idea that PD is a pathogenically heterogeneous state (Infante et al. 2016). Nowadays, there is pressing demand for the development of effective drugs for PD and the option of using LRRK2 kinase inhibitors is looked as a promising approach. However, there are some critical issues that need to be taken care off while employing LRRK2 kinase inhibitors in clinical practice, such as (I) there must be evaluation of the effect of LRRK2 kinase inhibition on its expression, (II) researchers must know whether the inhibition of LRRK2 will affect its non-kinase function (e.g. the interaction of LRRK2 with microtubule) and (III) does LRRK2 has a self-regulatory function associated with its autophosphorylation event. Various compounds with LRRK2 inhibitory activity have been developed (Taymansa and Greggio 2016). First-generation LRRK2 inhibitors such as LRRK2-IN-1, CZC-25146 and CZC-54252 had good pharmacokinetic profile but they could not penetrate blood–brain barrier. Additionally, LRRK2-IN-1 also had some off-target effects relating to inflammatory pathways. Second-generation LRRK2 inhibitors can cross the blood–brain barrier and include drugs like HG-10-102-1, GSK2578215A, TAE684, JH-II-127, GNE-7915, GNE-0877, GNE-9605, PF-06447475 and MLi-2. Various second-generation LRRK2 inhibitors notably GNE-0877, GNE-7915 and GNE-9605 have good pharmacokinetic properties and reasonable half-life, but they show side effect like lung abnormalities. Among numerous LRRK2 kinase inhibitors developed so far, PF-06447475 has promising features since as it has been tested in various mammalian species and shows least side effects. The major disadvantage of this compound is its variable half-life in different species (Chan and Tan 2017). We expect better LRRK2 inhibitors with better pharmacokinetic/clinical profile will be developed in the near future.
Creutzfeldt Jakob Disease
Creutzfeldt-Jakob disease (CJD) is a fatal neurodegenerative disease sometimes known as human form of mad cow disease. This disease is caused due to prion misfolding. Prion, a membrane anchored glycoprotein normally exists in the neurons of central nervous system (CNS) and is believed to affect several signalling pathways causing neurodegeneration that leads to spongiform appearance in the affected brain (Sattar 2013). Mutations in the prion protein encoding gene “PrP” located on chromosome 20p13 is considered one of the main reasons for altered prion folding, leading to CJD (Peoc’h et al. 2000). CJD prion is dangerous because it initiates a cyclic conversion of native prion protein into the diseased state. Once misfolded, the prion proteins interact with normally folded prions, also changing them to misfolded structures thereby complicating the disease furthermore (Clarke et al. 2001). Misfolded prion proteins are highly resistant to proteolytic cleavage and get aggregated in the brain causing damage to neurons (Prusiner et al. 1998). There occurs an exponential increase in the number of misfolded proteins in neurons and their aggregation forms insoluble CJD protein clumps leading to severe damage to neurons (Fig. 10). Accumulated CJD prions destroy brain by developing tiny holes in it thereby changing its shape to “spongy form”. A CJD patient often goes into coma and dies within a short span of 1–2 years. CJD is the most common human prion disease and is initially characterised by symptoms like dementia, personality changes, hallucinations, psychosis, paranoia and obsessive compulsive symptoms (Murray et al. 2012). Currently, there is no cure for this disease. Actual cause of the disease is still hidden from the eye of scientific community. However, researchers believe that mutations in the gene encoding prion protein can lead to misfolding of dominant alpha helical regions into beta pleated sheets thus introducing conformational changes in the prion protein. The defective prion proteins invade the brain and induce other prion protein molecules to misfold in a self-sustaining feedback loop. CJD has three forms, first one “acquired” i.e. transmissible from an affected person to a healthy person via exposure to brain or nervous-system tissue, mainly through certain medical procedures. In fact iatrogenic CJD can be transmitted by grafting infected tissue from a person who is either ill with the disease or in incubation. For example, some cases of dura matter grafts, a few exploratory neurosurgical interventions (stereo-EEG electrodes previously done in patients with CJD) and a case of corneal graft, are already on record (Fantini and Yahi 2015). CJD is transmitted through contact with tissues from organs like brain, eye (cornea), lung, kidney, liver, lymph, spleen or CSF and not through contact with blood (Scott 2013). Second form of CJD is the “sporadic form” that occurs without any known cause. By far sporadic form is the most common type of CJD and accounts for at least 85 percent of total cases. Sporadic CJD (sCJD) is rarely found in juvenile patients, and it usually affects adults in their 60s (De-Villemeur 2013). sCJD patients have a very short survival i.e. just 6 months. In fact, more than 90% of patients suffering from sCJD die within a year of symptom onset. The peak incidence of sCJD is in the 7th decade, while the disease shows less incidence in younger (20–40 s) or older (> 80) cases. As per the hypothesis, sCJD results either from the random structural change in the PrP protein causing the formation of PrPSc, or it results from a somatic prion protein (PRNP) gene mutation. Clinically, sCJD is characterised by features like ataxia, rapid cognitive decline, and myoclonus terminating in an akinetic mute state. The incidence rate of sCJD is reported as one per million populations per annum. However, mortality rates have gradually increased in the UK and in many other countries (Mackenzie and Will 2017). The third form of CJD is “hereditary form” where a person has a familial history of disease. If there is alteration in prion protein gene in a person’s sperm or egg cells, the mutation can be transmitted to the person’s offspring and these mutations in the prion gene are inherited as dominant traits. Variant CJD (vCJD), the novel human prion disease is a spontaneous neurodegenerative disease which occurs predominantly in the United Kingdom (UK). It has been associated to the consumption of beef products contaminated with the agent of the cattle disease, bovine spongiform encephalopathy (BSE) (Will et al. 2000; Will 2003; Ward et al. 2006). Moreover, it is now confirmed that that the secondary transmission of vCJD occurs through blood transfusion as seen in three clinical cases of vCJD linked to the transfusion of non-leucodepleted erythrocytes derived from individuals who themselves went on to develop VCJD (Urwin et al. 2016). Also, a recipient of an implicated transfusion who died of a non-neurological disorder was found to have PrPSc positivity in the spleen, indicating preclinical infection (Peden et al. 2004, Mackenzie and Will 2017). The field of CJD diagnosis has improved to a great extent with the advent of improved brain imaging and the potential for diagnostic tests in plasma and urine in vCJD and also the development of specific CSF tests in sCJD.

Pathophysiology of Creutzfeldt Jakob disease (CJD): Factors like mutations in prion gene, infections of nervous tissue and heredity transmission of infected prions can change the normal prion to infected/misfolded prion. Accumulation of infected/misfolded prion in the brain causes neurodegeneration characterised by spongiform brain with clinical manifestations like paranoia, dementia and obsessive compulsiveness
Targets for Creutzfeldt Jakob Disease
Theoretically, there are many possible targets for blocking CJD which include blocking conversion of PrPn to PrPres, stopping prion progression in secondary lymphoid organs, accelerating the clearance of amyloid PrP deposits in peripheral tissues and brain, promoting neuronal healing and reducing brain inflammation. Current therapeutic approaches rely mostly on blocking the conversion of PrP proteins to PrPres form for preventing the subsequent accumulation of PrPres in the central nervous system and peripheral nervous system. Based on these lines, recent targets include direct inhibition of this conversion, altering PrPn expression, blocking its cell surface localization, degradation of PrPres and interference with vital accessory molecules like glycosaminoglycans and fragment antigen binding. Factors like exposure to biomaterials contaminated with PrPres and mutations in PrP gene are believed as important causal agents of prion disease (Tamgüney et al. 2008). CJD can be dealt with the drugs inhibiting the aggregation of prion proteins in brain. Many such drugs have been developed, for example, pentosan sulphate, vidarabine, quinacrine, amantadine, acyclovir, curcumin, doxycycline, astemizole and flupirtine. These drugs have the ability to reduce the symptoms of familial CJD. Aggregation of prion proteins in the body does not trigger any immune response since prion proteins exist normally in the body and it is the point mutation that generates abnormal prion resistant to protease. Strategies like targeting with miRNA, gene silencing and antibodies can serve better to tame this disease (Linden et al. 2008; Manuelidis 2013). Targeting laminin, a high affinity prion receptor (LPR/LR) is regarded as a potential candidate for CJD therapy. Studies indicate that LRP/LR acts as a receptor for both the normal (PrPn) as well as the diseased form of prion (PrPres) (Gauczynski et al. 2006). This receptor is believed to play crucial role in Prpres binding and its intracellular internalisation. Gauczynski and co-workers reported that polysulfated glycans can block the synthesis of abnormal PrP by inhibiting of LPR/LR-dependent binding of prions to target cells (Gauczynski et al. 2006; Mbazima et al. 2010). In an another experiment, Leucht and co-workers (Leucht et al. 2004) employed antisense RNA to knockdown the LPR/LR in mouse brain so that such animals could serve as experimental tools for elucidating the role of LRP/LR in pathogenesis of prion diseases. In another study using lentiviral vectors to generate small interfering RNA (siRNAs) against the laminin receptor precursor mRNA, there was a prolonged extension in the preclinical phase of scrapie (a prion disease)-infected mice. Moreover, stereotactic intra-cerebral microinjection of recombinant lentiviral vectors expressing siRNA to LRP7 and LRP9 into the hippocampus was found to effectively extend the preclinical phase in scrapie-infected mice (Pflanz et al. 2009). More research is needed to identify the ligands/compounds targeting this receptor so as to tame CJD. Various other studies have tried to block prp, for example, RNAi-mediated knockdown of PrP and the resultant inhibition of PrPSc (Tilly et al. 2003). Virally expressed RNAi has been employed for reducing the levels of PrPC in goats, cattle and mice (Golding et al. 2006; White and Mallucci 2009). Transgenic mice produced by lentiviral transduction of embryos stably express anti-PrP shRNAs and have increased resistance to prion infection due to resultant RNAi of endogenous PrP (Pfeifer et al. 2006). The major hindrance to the use of therapeutic RNAi in taming neurodegenerative disease is its delivery to the brain. Blood–brain barrier (BBB) restricts the passive entry of molecules from the peripheral circulation thus forcing researchers to use alternative ways for delivering RNAi into brain like the transient disruption of the BBB’s impermeability, delivery through active transport (across this barrier), or direct injection into the brain. Various new technologies have recently been recently developed which include utilising viruses capable of transversing the BBB (unaided) or targeting receptors in the BBB to mediate the transport of RNAi (Pfeifer et al. 2006). Transcription factors namely SP1 and MTF-1 have been found to significantly increase prion protein levels and upregulate prion gene expression in copper-replete Menkes protein (MNK) deletion cells. Also, siRNA-mediated “knockdown” of SP1 or MTF-1 in MNK deletion cells reduces prion protein levels and down-regulates prion gene expression. These results support a novel mechanism through which SP1 and MTF-1 act as copper-sensing transcriptional activators to regulate human prion gene expression (Bellingham et al. 2009; Panegyres and Armari 2013). Therefore, SP1 and MTF-1 represent the new targets for developing key therapeutics to modulate the expression of the cellular prion protein and subsequently the prevention of prion disease like CJD.
Anti-prion antibodies directed against PrPC represent another strategy to treat CJD. However, this strategy is still in its infancy and needs more advancement. Earlier, monoclonal antibody like D13 was reported to exhibit strong target-related toxicity. Yet another antibody ICSM18, with an epitope that overlaps with antibody POM1, was reported to be safe when injected into brains of mouse. However, the subsequent studies contradicted these findings. In fact several antibodies targeted against certain epitopes of PrPC, including antibody POM1, were found extremely neurotoxic. When assessed with dose-escalation studies using diffusion-weighted magnetic resonance imaging and various histological techniques, Reimann and co-workers found that both D13 and ICSM18 induce rapid, dose-dependent, on-target neurotoxicity. They concluded that antibodies directed to this region may not be suitable as therapeutics. However, they also reported that no such toxicity was observed when antibodies were administered against the flexible tail present at amino-terminus of the prion protein (Reimann et al. 2016), thus suggesting that any attempt aimed at immunotherapy of prion diseases should take into account such potential untoward effects/threats.
Targeting Mitochondrial Proteins in Neurodegeneration
Human brain requires abundant energy supply and utilises about 20% of total bodily consumption of oxygen for its normal functioning. Mitochondria being the power house generate this huge energy through the process called oxidative phosphorylation (OXPHOS) (Moreira et al. 2009). Major portion of this energy is used to maintain ion gradients across the plasma membranes of neurons for generating action potentials. Making neurons deficient of oxygen or glucose even for a brief interval of time may result in neuronal death. OXPHOS although a necessary process for energy generation, also produces various toxic by-products in the form of free radicals like superoxide (O2−), hydrogen peroxide (H2O2) and hydroxyl (OH−). These free radicals could damage various cellular components if not cleared immediately. Various mitochondrial enzymes/proteins detoxify such free radicals, for instance O2− is detoxified by the mitochondrial manganese superoxide dismutase (MnSOD) producing H2O2 which in turn can be converted to H2O by another enzyme–glutathione peroxidase (GPx) (Moreira et al. 2010). Deregulation in the normal expression of such crucial enzymes can drastically elevate the levels of reactive oxygen species thereby damaging neurons and predisposing them to neurodegeneration Mitochondrial electron transport chain (ETC) is composed of various enzyme complexes namely complex-I (NADH-dehydrogenase), complex-II (succinate-dehydrogenase), complex-III (ubiquinol-cytochrome-C-oxidoreductase), complex-IV (cytochrome-C-oxidase) and complex-V (ATP-synthase) (Coussee et al. 2011). Deficiency of these mitochondrial complexes/enzyme systems has clear association with the pathogenesis of neurodegenerative diseases like PD, AD, HD and ALS.
Reports suggest that increase in the expression of hyperphosphorylated tau and Aβ plaques, mitochondrial α-synuclein, mitochondrial Htt and mitochondrial SOD1 lead to combined deficiencies of mitochondrial complexes (Mojtaba et al. 2017) thus promoting the accumulation of aggregated/misfolded form of such proteins through the inhibition of the proteasome activity during AD, PD, HD and ALS, respectively (Fig. 11). The mechanism of mitochondrial dysfunction in Alzheimer’s disease shows the involvement of various proteins/enzymes. Initially, Aβ precursor protein (APP) blocks the translocase enzyme of mitochondrial outer membrane (TOM40), and inner membrane (TIM23) thus blocking the entry of nuclear-encoded subunits of COX/CIV into mitochondria and declining cytochrome-oxidase (COX/CIV) activity. Alternatively, Aβ can enter the mitochondria through TOM complex, interact with the heme groups of COX/CIV, decrease its catalytic activity and thus elevate ROS production which in turn elevates Aβ production as well as cyclophilin D-mediated (CypD) opening of permeability transition pore (PTP). This is followed by the release of pro-apoptotic factors such as cytochrome c (Cyt c) and apoptosis-inducing factor (AIF) through PTP into the cytosol where it activates the apoptotic pathway thus leading to death of cells/neurons (Hibiki and Giovanni 2017). In case of PD, mitochondrial dysfunction has been associated with inhibition of mitochondrial complex-I (CI) activity. Mutations in parkin and tensin homologue (PTEN)-induced kinase 1 (PINK1) involved in mitochondrial function are considered as the genetic causes of PD. PINK1 causes phosphorylation of parkin on threonine 175 thus eliciting its translocation to mitochondria. Overexpression of PINK1 seems to protect mitochondria by inhibiting release of cytochrome c (Cyt c) from mitochondria, increasing expression of mitochondrial CI subunits, and reducing reactive oxygen species production. All such beneficial effects are found lost in parkin-deficient experimental models which show decline in CI and complex-IV (CIV) subunits and an increase in ROS production. ROS generation potentiates the cyclophilin D (CypD)-mediated permeability transition pore (PTP) opening and consequent release of Cyt c from mitochondria thus promoting apoptotic pathways and cell death (Moreira et al. 2010). Therefore, strategies to increase ETC capacity or to reduce mitochondrial ROS production by modulating expression of proteins like parkin, PINKY, Cyt c and mitochondrial CI subunits may prove useful for the treatment of brain ageing and neurodegenerative diseases.

Proteinopathy triggered mitochondrial complex deficiencies during neurodegenerative disorders like amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), Alzheimer’s disease (AD) and Parkinson’s disease (PD)
Mitophagy, an important phenomenon associated with neurodegenerative diseases refers to the selective degradation of mitochondria due to autophagy. Compromised mitophagy lead to the accumulation of protein aggregates which ultimately conclude with neurodegeneration. In mammals, mitophagy is activated by ROS generation and mitochondrial permeability transition pore (PTP) induction. One of the most studied pathways for mitophagy in mammalian cells is the PINK1/Parkin pathway (Youle and Narendra 2011; Pickrell and Youle 2015). In this pathway, mitochondrial damage decreases the mitochondrial membrane potential which leads to the stabilisation of the ubiquitin kinase (PTEN)-induced kinase 1 (PINK1) on the outer mitochondrial membrane. Here it phosphorylates ubiquitin, thus leading to the recruitment of the E3 ubiquitin ligase parkin. PINK1 phosphorylation activates parkin which polyubiquitinates mitochondrial proteins leading to their association with the ubiquitin-binding domains of autophagy receptors and the formation of the autophagosome. The autophagosome then fuses with the lysosome, leading to degradation of the mitochondria (Lazarou et al. 2015). Alternatively, PINK1 can recruit autophagy receptors directly in a parkin-independent manner, leading to low levels of mitophagy. Parkin-mediated mitophagy can be suppressed by de-ubiquitination of its substrates. USP8 (Ubiquitin-Specific Peptidase 8) was found to regulate mitophagy in human cell lines (Durcan et al. 2014), while the reduction of USP30 (Bingol et al. 2014) and USP15 (Cornelissen et al. 2014) can increase mitophagy and rescue mitochondrial phenotypes in fly models. Mutations in Parkin (Kitada et al. 1998) and PINK1 (Valente et al. 2004) lead to the autosomal recessive form of Parkinson’s disease. Growing number of studies indicate the protective role of mitophagy in several deleterious situations such as CoQ10 deficiency (Gegg et al. 2009), hypoxia (Zhang et al. 2008) and rotenone exposure (Pan et al. 2009) thereby making mitophagy and the associated proteins a good candidate(s) for therapeutic intervention.
Parkin being a possible link between mitochondrial dysfunction, mitochondrial fission and mitophagy during PD plays crucial role in the turnover of damaged mitochondria by mitophagy (McBride 2008) rendering this protein a potential target for therapeutic intervention to improve mitophagy during PD. Studies also show that Parkin is selectively recruited to dysfunctional mitochondria promoting their fragmentation and destruction in the autophagosomes (Narendra et al. 2008). These results suggest that mutations of parkin can impair mitophagy which in turn can enhance the accumulation of dysfunctional mitochondria. Thus, targeting mitophagy-related proteins in neurodegenerative disorders may open the avenues for potential targets for discovering effective therapies.
Mitochondrial biogenesis is also seen as an important factor linked to neurodegeneration. It is associated with expression of genes including peroxisome proliferator-activated receptor-c coactivator 1 alpha (PGC-1α), mitochondrial transcription factor A (TFAM), nuclear respiratory factor 1 and 2 (NRF1 and NRF2), and mitochondrial transcription factor B1 (TFB1 M) (Golpich et al. 2015). It is highlighted that impaired mitochondrial biogenesis potentially contributes to the mitochondrial dysfunction and neurodegenerative diseases. Evidences suggest the possible role of various signal transduction proteins, transcription factors and transcription co-activators in the regulation of mitochondrial mass and number inside neurons (Onyango et al. 2010; Dominy and Puigserver 2013). Mitochondrial biogenesis is dependent on several signalling cascades and transcriptional complexes that promote the formation and assembly of mitochondria. It is a process that is heavily dependent on timely and coordinated transcriptional control of genes encoding for mitochondrial proteins.
PGC-1 family members (e.g. PGC-1α and PGC-1β) coactivate genes encoding proteins for transcription and replication of mtDNA as well as importation of mitochondrial protein (Wu et al. 2006; Mojtaba et al. 2017). They also have contribution in the physiological integration of mitochondrial biogenesis with oxidative metabolism and provide an overlapping and amplifying regulation of several nuclear-encoded mitochondrial genes (Scarpulla 2011). As a co-transcriptional regulation factor, the PGC-1α provokes mitochondrial biogenesis by activating several transcription factors, including NRF1 and NRF2. Furthermore, it is also called GA-binding protein A (GABPA) that regulates the expression of multiple nuclear genes to encode mitochondrial proteins such as TFAM. The TFAM in turn contributes to the mtDNA maintenance as well as motivates the replication and transcription of both mtDNA and PPARs (Scarpulla 2008; Yin et al. 2008; Sharma et al. 2014). As a transcriptional coactivator, PGC-1 functions together with combination of other transcription factors such as peroxisome proliferator-activated receptors (PPARs) in the regulation of mitochondrial biogenesis. PPARs, in particular PPAR-c, may be a major signalling pathway involved in neuroinflammation. PPAR-c is also an important regulatory factor in the modulation of genes having PPAR response element (PPRE) in their promoters, such as those encoding for oxidative stress, inflammation (COX-2), inducible nitric oxide synthase (iNOS), nuclear factor-kappaB (NF-kB), and apoptosis (Kiaei 2008). In addition, mitochondrial biogenesis involves transcription factors such as sirtuins. Sirtuins (SIRTs) are a family of the nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases which contribute in cellular processes such as cell cycle, transcription, energy metabolism, mitochondrial functions, ageing, apoptosis and cell survival. Mammals possess seven sirtuins with different activities, localised to the nucleus (SIRT1, SIRT6 and SIRT7), cytosol (SIRT2) and mitochondria (SIRT3, SIRT4 and SIRT5). Sirtuins can influence the progression of neurodegenerative disorders by modulating transcription factor activity (Kanfi et al. 2012; Johri and Beal 2012; Min et al. 2013; Herskovits and Guarente 2013). Sirtuins as the transcriptional regulators may have potential therapeutic effects on a several chronic age-related and aggregate-forming neurodegenerative diseases including AD, PD, HD and ALS.
Antioxidant peptides like SS31 and SS20 represent a novel therapeutic approach that can scavenge mitochondrial free radicals and also inhibit mitochondrial permeability transition and cytochrome c release which prevents oxidant-induced cell death (Cho et al. 2007; Johri and Beal 2012; Hou et al. 2016;). Recently, preclinical studies support potential use of the mitochondria-targeted antioxidants as an effective treatment for neurodegenerative disorders (Szeto 2008). Manczak and her colleagues stated that SS31 prevents Aβ toxicity as well as decreases learning and memory deficits (Manczak et al. 2010). Moreover, SS31 and SS20 demonstrated significant neuroprotective effects on dopaminergic neurons of MPTP-treated mice (Yang et al. 2009). SS31 may also be used to treat HD due to its property to promote mitochondrial function and neuronal viability (Chandra et al. 2014). SS31 is considered a novel therapeutic approach to treat neuronal damage induced by oxidative stress. It targets the ROS production at the inner mitochondrial membrane and prevents further mitochondrial damage (Petri et al. 2006). Taken together, proteins involved in mitochondrial ETC functioning, mitophagy and biogenesis represent a novel therapeutic target and confirm a modern neuroprotective approach for most of diseases such as AD, PD, HD and ALS in the near future.
Oxidative Damage, Proteinopathy and Neurodegeneration
Studies with transgenic animal models reveal that oxidative modifications of brain proteins result in their functional loss and hence neurodegeneration. Reactive oxygen species cause proteotoxicity and neurotoxicity mostly via three major mechanisms i.e. (1) impairing proteasomal system, (2) damaging lysosomes and (3) promoting Aβ and α-synuclein oligomerization (Fig. 12).

Oxidative stress, free radical generation and neuronal damage
Proteasomal system especially the 26S proteasome represents one of the pivotal cellular mechanisms damaged by oxidative modifications. Impaired proteasomal system prevents the degradation of toxic protein aggregates (Aiken et al. 2011; Pajares et al. 2015). Excessive accumulation of mutant or wild-type proteins inside cells triggers conformational changes in proteins (e.g. helix to β-strand conformation) which promote more self-aggregation and loss of function. Heat shock proteins like Hsp40, Hsp70, Hsp90 and others normally avert misfolding of intracellular proteins but their excess accumulation or redox modifications may disrupt the normal expression of proteins and inflict toxic effects on various cellular organelles including lysosomes and mitochondria (Morimoto 2008; Niforou et al. 2014).
Lysosomes play critical role in the removal of excessive/toxic proteins from a cell. Their destruction due to ROS promotes intracellular accumulation of several proteins (Pajares et al. 2015). For instance, oligomerisation of Aβ and α-synuclein leads to proteotoxicity (Snead and Eliezer 2014; Xiang et al. 2015). The proteotoxicity in turn causes impairment of mitochondrial function. Studies based on analysis of postmortem brain samples, isolated mitochondria and cultured cells suggest that oligomerised proteins or even the monomers of α-synuclein or Aβ can impair mitochondria by altering mitochondrial fusion/fission processes, mitophagy and bioenergetic (Devi et al. 2006; Wang et al. 2009; Banerjee et al. 2010; Protter et al. 2012). The fact that Aβ production triggers ROS generation has been proved by various invivo studies, cell-free chemical systems and cell cultured experiments. Both Aβ40 and Aβ42 can interact with transition elements via different amino acid residues including His14, His13 and His6. These bound elements can participate in various redox-cycling reactions thus producing ROS (Smith et al. 2007). Postmortem evidences show the presence of elevated levels of metals like Zn, Fe and Cu in the brain of AD patients. These metals are located mainly near the plaque which suggests the putative role of Aβ-mediated ROS production in AD pathogenesis.
Aβ may also increase intracellular ROS production via the involvement of apoptosis signal-regulating kinase 1 (ASK1) (Kadowaki et al. 2005a, b). Neuronal cell culture experiments show that Aβ promotes ROS production seemingly through the activation of reduced NADPH oxidase (Shelat et al. 2008; Hu and Li 2016). The other mechanism through which Aβ induces ROS production includes microglial activation via fibrillar Aβ as observed in microglial cell cultures and coculture of microglia and neurons (Qin et al. 2002; Qin et al. 2006). Microglial cells possess a B-type of scavenger receptor known as CD36. Fibrillar Aβ acts as a ligand and binds CD36 to stimulate microglial ROS production, cytokine expression (such as IL-6, IL-1β, TNF)-α) and phagocytosis in AD brain (Coraci et al. 2002; Doens and Fernández 2014).
PD brain contains elevated levels of oxidative damage markers and transition metals such as Fe (Jenner and Olanow 1996; Sofic et al. 1988). Increased expression of α-synuclein during PD could presumably enhance the production of ROS as observed in several experimental models (Bir et al. 2014; Di-Maio et al. 2016). Overexpression of mutant or wild α-synuclein has been found to be associated with elevation in the levels of ROS in SH-SY5Y cells (Anandhan et al. 2015). Studies based on biophysical analysis indicate the binding of elements like Cu and Fe to α-synuclein that may increase the cytotoxicity of α-synuclein (Carboni and Lingor 2015). Iron which promotes ROS production and catalyses peroxidative damage to biomolecules also causes translational upregulation of α-synuclein (Febbraro et al. 2012) in PD brains. Fe can prompt the formation of large oligomers of α-synuclein that can form membrane-spanning channels and thus lead to intracellular toxicity. Interaction of Fe with α-synuclein may even trigger the formation of H2O2 through various redox reactions (Carboni and Lingor 2015).
Since PD involves loss of DAergic neurons, it is believed that the oxidation products of DA notably the toxic quinines and ROS could act as contributing factors in the pathology of PD (Hastings 2009; Jana et al. 2011). DA oxidation products seem to have a modulatory effect on the oligomerisation and cytotoxicity of α-synuclein (Leong et al. 2009). Rat DAergic cell lines and human DAergic neurons when exposed to paraquat lead to increased aggregation of α-synuclein and this aggregation is critically dependent on NADPH oxidase, thus giving a clear indication that ROS has an important role in PD pathology. There also occurs gathering of oxidative damage markers in the substantia nigra, which can be eliminated by using adeno-associated virus-mediated overexpression of a specific shRNA to knockdown NADPH oxidase (Nox1) gene (Cristóvão et al. 2012).
Superoxide dismutase (SOD) is an important free radical scavenger/antioxidant enzyme. Earlier reports suggest an increased expression of SOD1 immunoreactivity in Bergmann glia and astrocytes in cerebellum of CJD cases. Also there was an elevated expression of SOD1 in the cerebral cortex of CJD cases (Freixes et al. 2006). Recently, Brazier and his colleagues used an agent with actions similar to SOD in a rodent model which extended the life of scrapie-infected animals thus raising the possibility of using free radical scavengers and antioxidants as the possible therapeutics in the treatment of prion diseases. To provide further insights into the relative pathogenic importance of oxidative stress, Brazier and his colleagues used a potent manganese-superoxide dismutase/catalase mimetic, EUK-189, as a therapeutic agent in the mouse model of human prion disease. They observed a significant but relatively modest prolongation of survival due to EUK-189-treatment, which correlated with reductions in ROS and reactive nitrogen species (RNS) mediated damage to proteins when compared to untreated disease controls. In addition, EUK-189 also caused reductions in spongiform change in specific brain regions of terminally sick mice as confirmed by lesion profiling (Brazier et al. 2008). These results suggest a prominent role of antioxidants in ameliorating/modulating prion diseases. (nbr2). There also occurs decreased expression of nNOS along with its misfolding and abnormal localization in neuroblastoma cells as well as the brain during scrapie infection (Ovadia et al. 1996; Keshet et al. 1999; Petersen et al. 2005). Furthermore, reports indicate increased levels of inducible nitric oxide synthase and endothelial and in the cerebral cortex in sporadic CJD (Freixes et al. 2006). Thus, oxidative stress-mediated derangements of brain proteins/enzymes have deeper implications in the pathology of neurodegenerative diseases and could serve as premier source for targeting of drugs.
Conclusion
Neurodegenerative diseases have complex pathophysiology and result due to multiple proteinopathies as discussed in this review. Treating damaged neurons poses perplexing challenges notably the blood–brain barrier, non-specificity/off-targets, side effects and weak pharmacokinetic profile of proposed drugs. Research should focus on developing therapeutics that are target specific, have ability to cross blood–brain barrier, possess least possible side effects and show cost effectiveness. Since many neurodegenerative diseases involve cross talk or co-existence of several proteinopathies therefore designing drugs with ability to strike multiple targets in the pathophysiology of ND’s seems to be an effective strategy. Interactions between synucleinopathy and tauopathy can promote neurodegeneration via distinct mechanisms such as (I) α-Synuclein can bind directly to tau and tubulin protein and thus block the normal interaction between the two proteins thus interfering with their physiological function, (II) α-Synuclein could promote hyperphosphorylation of tau protein by recruiting various kinases, (III) α-Synuclein can promote tau aggregation and the formation of tau aggregates, (IV) α-synuclein oligomers may in turn seed tau fibrillation and formation of insoluble NFTs thus triggering tauopathy. Therapeutics including synthetic chaperones, kinase inhibitors, anti-aggregation agents, antibodies, LRRK2 inhibitors, mAChRs activators, ApoE4 inhibitors, dopaminergic and NAergic drugs could prove effective medicines in taming various ND’s; however, there is need for shift from ‘magic bullet’ to ‘magic shotgun’ strategy. While combing several drug entities that act independently on different etiological targets of a disease researches must take utmost care of difference in (1) bioavailability of each individual entity of a single drug, (2) pharmacokinetic parameters, (3) metabolic profile and overall (4) the unpredicted drug–drug interactions that will result in the development of ideally effective drugs. Prospective strategies need to focus on combinatorial progress and advancement in neuro-proteome-based therapeutic regimen, clinical data, bioinformatics and neuro-imaging technology that can verily improve the diagnosis, prognosis, as well as the therapeutic outcome of neurodegenerative diseases.
References
Abreu PA, Castro HC, Paes-de-Carvalho R et al (2013) Molecular modeling of a phenylamidine class of NMDA receptor antagonists and the rational design of new triazolyl-amidine derivatives. Chem Biol Drug Des 81:185–197
Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J (2012) An effector- reduced anti- β-amyloid (Aβ) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci 32:9677–9789
Agrawal M, Biswas A (2015) Molecular diagnostics of neurodegenerative disorders. Front Mol Biosci 2:54
Aiken CT, Kaake RM, Wang X, Huang L (2011) Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteom 10:R110006924
Alasmari F, Alshammari MA, Alasmari AF, Alanazi WA, Alhazzani K (2018) Neuroinflammatory cytokines induce amyloid beta neurotoxicity through modulating amyloid precursor protein levels/metabolism. Biomed Res Int 2018:1–8
Alonso ADC, Zaidi T, Novak M, Barra HS, Grundke-Iqbal I, Iqbal K (2001) Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J Biol Chem 276:37967–37973
Anandhan A, Rodriguez-Rocha H, Bohovych I et al (2015) Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol Dis 81:76–92
Ansah TA, Ferguson MC, Nayyar T, Deutch AY (2011) Age and durationdependent effects of MPTP on cortical serotonin systems. Neurosci Lett 504(2):160–164
Arima K, Hirai S, Sunohara N et al (1999) Cellular co-localization of phosphorylated tau-and NACP/α-synuclein-epitopes in lewy bodies in sporadic Parkinson’s disease and in dementia with lewy bodies. Brain Res 843:53–61
Arndt JW, Qian F, Smith BA, Quan C, Kilambi KP, Bush MW, Walz T, Pepinsky RB, Bussière T, Hamann S, Cameron TO (2018) Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid- β. Sci Rep 8:6412
Asaoka Y, Nakamura S, Yoshida K, Nishizuka Y (1992) Protein kinase C calcium and phospholipi degradation. Trends Biochem 17:414–417
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a drosophila model for Parkinson’s disease. Science 295:865–868
Banerjee K, Sinha M, Pham CLL et al (2010) Alpha-synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: implications in Parkinson’s disease. FEBS Lett 584(8):1571–1576
Bara-Jimenez W, Sherzai A, Dimitrova T, Favit A, Bibbiani F, Gillespie M, Morris JM, Mouradian MM, Chase TN (2003) Adenosine A2A receptor antagonist treatment of Parkinson’s disease. Neurology 61:293–296
Barthélemy NR, Gabelle A, Hirtz C, Fenaille F, Sergeant N, Schraen-Maschke S, Vialaret J, Buée L, Junot C, Becher F, Lehmann S (2016) Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer’s disease, progressive supranuclear palsy, and dementia with lewy bodies. J Alzheimers Dis 51:1033–1043
Bazan NG (1999) Eicosanoids, platelet-activating factor and inflammation. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD (eds) Basic neurochemistry. Raven Press Ltd, New York
Bellingham SA, Coleman LA, Masters CL, Ca-makaris J, Hill AF (2009) Regulation of prion gene expression by transcription factors SP1 and metal transcription factor-1. J Biol Chem 284:1291–1301
Bespalov MM, Saarma M (2007) GDNF family receptor complexes are emerging drug targets. Trends Pharm Sci 28(2):68–74
Bespalov MM, Sidorova YA, Suleymanova I et al (2016) Novel agonist of GDNF family ligand receptor RET for the treatment of experimental neuropathy. BioRxiv 1:061820
Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, Ganie SA (2015) Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases, a mechanistic insight. Biomed Pharmacother 74:101–110
Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510:370–375
Bir A, Sen O, Anand S et al (2014) α-Synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: implications in the pathogenesis of Parkinson’s disease. J Neurochem 131:868–877
Bohrmann B, Baumann K, Benz J et al (2012) Gantenerumab: a novel human anti- Aβ antibody demonstrates sustained cerebral amyloid- β binding and elicits cell- mediated removal of human amyloid- β. J Alzheimers Dis 28:49–69
Bosco D, Fava A, Plastino M, Montalcini T, Pujia A (2011) Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J Cell Mol Med 15:1807–1821
Bouter Y, Noguerola JS, Tucholla P, Crespi GA, Parker MW, Wiltfang J, Miles LA, Bayer TA (2015) Aβ targets of the biosimilar antibodies of bapineuzumab, crenezumab, solanezumab in comparison to an antibody against N- truncated Aβ in sporadic Alzheimer disease cases and mouse models. Acta Neuropathol 130:713–729
Brash AR (1999) Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem 274(34):23679–23682
Brazier MW, Doctrow SR, Masters CL, Collins SJ (2008) A manganese-superoxide dismutase/catalase mimetic extends survival in a mouse model of human prion disease. Free Radic Biol Med 45(2):184–192
Bruinsma IB, Bruggink KA, Kinast K (2011) Inhibition of alpha-synuclein aggregation by small heat shock proteins. Proteins 79:2956–2967
Carboni E, Lingor P (2015) Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 7(3):395–404
Caulfield MP, Bridsall NJ (1998) International Union of Pharmacology XVII classification of muscarinic acetylcholine receptors. Pharmacol Rev 50:279–290
Chakrabarti S, Khemka VK, Banerjee A, Chatterjee G, Ganguly A, Biswas A (2015) Metabolic risk factors of sporadic Alzheimer’s disease: implications in the pathology, pathogenesis and treatment. Aging Dis 6(4):282–299
Chan SL, Tan EK (2017) Targeting LRRK2 in Parkinson’s disease: an update on recent developments. Expert Opin Ther Targets 21(6):601–610
Chandra A, Johri A, Beal MF (2014) Prospects for neuroprotective therapies in prodromal Huntington’s disease. Mov Disord 29:285–293
Chen JF, Xu K, Petzer JP et al (2001) Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci 21:RC143
Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT (2007) A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem 282:4634–4642
Chow VW, Mattson MP, Wong PC, Gleichmann M (2010) An overview of APP processing enzymes and products. NeuroMol Med 121:1–12
Clarke AR, Jackson GS, Collinge J (2001) The molecular biology of prion propagation. Philos Trans R Soc Lond, Ser B 356:185–195
Colell A, Fernandez A, Fernandez-Checa JC (2009) Mitochondria, cholesterol and amyloid β peptide: a dangerous trio in Alzheimer disease. J Bioenerg Biomembr 41:417–423
Colotta V, Lenzi O, Catarzi D et al (2012) 3-Hydroxy-1H-quinazoline-2,4-dione derivatives as new antagonists at ionotropic glutamate receptors: molecular modeling and pharmacological studies. Eur J Med Chem 54:470–482
Congdon EE, Krishnaswamy S, Sigurdsson EM (2014) Harnessing the immune system for treatment and detection of tau pathology. J Alzheimers Dis 40:S113–S121
Coraci IS, Husemann J, Berman JW et al (2002) CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol 160(1):101–112
Coric V, Van-Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW et al (2012) Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol 69:1430–1440
Cornelis J, Schyf VD (2011) the use of multi-target drugs in the treatment of neurodegenerative diseases. Exp Rev Clin Pharmacol 43:293–298
Cornelissen T, Haddad D, Wauters F, Van-Humbeeck C, Mandemakers W, Koentjoro B (2014) The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Human Mol Genet 23:5227–5242
Coussee E, De-Smet P, Bogaert E, Coussee E, De-Smet P, Bogaert E, Elens I, Van-Damme P, Willems P, Koopman W, Van-Den-Bosch L, Callewaert G (2011) G37R SOD1 mutant alters mitochondrial complex I activity, Ca2+ uptake and ATP production. Cell Calcium 49:217–225
Cox P, Davis D, Mash D, Metcalf J, Banack S (2016) Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proc R Soc B 283(1823):20152397
Cristalli G, Muller CE, Volpini R (2009) Recent developments in adenosine A2A receptor ligands. Handb Exp Pharmacol 193:59–98
Cristóvão AC, Guhathakurta S, Bok E et al (2012) NADPH oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J Neurosci 32(42):14465–14477
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295
D’Andrea MR, Nagele RG (2010) Morphologically distinct types of amyloid plaques point the way to a better understanding of Alzheimer’s disease pathogenesis. Biotech Histochem 85:133–147
Davie BJ, Christopoulos A, Scammells PJ (2013) Development of M1 mAChR allosteric and bitopic ligands: prospective therapeutics for the treatment of cognitive deficits. ACS Chem Neurosci 4(7):1026–1048
Dehay B, Bove J, Rodríguez-Muela N et al (2010) Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci 30:12535–12544
Dehay B, Bourdenx M, Gorry P, Przedborski S, Vila M, Hunot S, Singleton A, Olanow CW, Merchant KM, Bezard E, Petsko GA (2015) Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol 14:855–866
De-Lera-Ruiz M, Lim YH, Zheng J (2013) Adenosine A2A receptor as a drug discovery target. J Med Chem 57(9):3623–3650
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti- Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβburden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 98:8850–8855
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26(35):9057–9068
De-Villemeur TB (2013) Creutzfeldt-Jakob disease Handbook of clinical neurology. Elsevier 112:1191–1193
Di-Maio R, Barrett PJ, Hoffman EK et al (2016) α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med 8(342):342ra78
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7–61
Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nat Neurosci 5:452–457
Doder M, Rabiner EA, Turjanski N, Lees AJ, Brooks DJ (2003) Study CWP: tremor in Parkinson’s disease and serotonergic dysfunction: an 11C-WAY 100635 PET study. Neurology 60(4):601–605
Doens D, Fernández PL (2014) Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J Neuroinflammation 11:48
Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S et al (2005) Phenotypic and biochemical analyses of BACE1-and BACE2-deficient mice. J Biol Chem 280:30797–30806
Dominy JE, Puigserver P (2013) Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol 5:1–18
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S et al (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. New Engl J Med 369:341–350
Du X, Wang X, Geng M (2018) Alzheimer’s disease hypothesis and related therapies. Transl Neuro 7(1):2. https://doi.org/10.1186/s40035-018-0107-y
Durcan TM, Tang MY, Pérusse JR et al (2014) USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J 33:e201489729
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z et al (2011) Distinct roles in vivo for the ubiquitin–proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J Neurosci 3141:14508–14520
Ebrahimi-Fakhari D, Saidi LJ, Wahlster L (2013) Molecular chaperones and protein folding as therapeutic targets in Parkinson’s disease and other synucleinopathies. Acta Neuropathol Commun 1:79. https://doi.org/10.1186/2051-5960-1-79
Engelender S (2008) Ubiquitination of alpha synuclein and autophagy in Parkinson’s disease. Autophagy 4:372–374
Erin E, Congdon Einar MS (2018) Tau-targeting therapies for Alzheimer disease. Neurology. https://doi.org/10.1038/s41582-018-0013-z
Espinoza-Moraga M, Caballero J, Gaube F, Winckler T, Santos LS (2012) 1-Benzyl-1, 2, 3, 4-tetrahydro-b-carboline as channel blocker of N-methyl-D-aspartate receptors. Chem Biol Drug Des 79:594–599
Ezza HSA, Khadrawy YA (2014) Glutamate excitotoxicity and neurodegeneration. J Mol Genet Med 8:4
Fallon L, Belanger CM, Corera AT et al (2006) A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI 3 K-Akt signalling. Nat Cell Biol 8:834–842
Fantini J, Yahi N (2015) Brain lipids in synaptic function and neurological disease: clues to innovative therapeutic strategies for brain disorders. Academic Press, London
Febbraro F, Giorgi M, Caldarola S, Loreni F, Romero-ramos M (2012) α-Synuclein expression is modulated at the translational level by iron. NeuroReport 23(9):576–580
Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C et al (2009) Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry 48:10047–10055
Fiskum G, Starkov A, Polster BM, Chinopoulos C (2003) Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci 991:111–119
Fitzgerald DP, Emerson DL, Qian Y et al (2012) TPI-287, a new taxane family member reduces the brain metastatic colonization of breast cancer cells. Mol Cancer Ther 11:1959–1967
Folch J, Ettcheto M, Petrov D, Abad S, Pedros I, Marin M et al (2018) Review of the advances in treatment for Alzheimer disease: strategies for combating β-amyloid protein. Neurologia 33:47–58
Foley TD (1997) 5-HPETE is a potent inhibitor of neuronal Na K-ATPase activity. Biochem Biophys Res Commun 235(2):374–376
Foster DJ, Choi DL, Conn PJ, Rook JM (2014) Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsychiatr Dis Treat 10:183. https://doi.org/10.2147/NDTS55104
Francesco P, Madia L, Giancarlo L, Bruno PI (2019) A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. https://doi.org/10.1038/s41582-018-0116-6
Freixes M, Rodriguez A, Dalf´o E, Ferrer I (2006) Oxidation glycoxidation, lipoxidation, nitration, and responses to oxidative stress in the cerebral cortex in Creutzfeldt-Jakob disease. Neurobiol Aging 27:1807–1815
Fuller JP, Stavenhagen JB, Christensen S, Kartberg F, Glennie MJ, Teeling JL (2015) Comparing the efficacy and neuroinflammatory potential of three anti- Aβantibodies. Acta Neuropathol 130:699–711
Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM et al (1996) Increased activity-regulating and neuroprotective efficacy of α-secretase-derived secreted amyloid precursor protein conferred by a c-terminal heparin-binding domain. J Neurochem 67:1882–1896
Gagliardi RJ (2000) Neuroprotection, excitotoxicity and NMDA antagonists. Arq Neuro-Psiquiatr 58:583–588
Galpern WR, Lang AE (2006) Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol 59:449–458
Gauczynski S, Nikles D, El-Gogo S et al (2006) The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J Infect Dis 194:702–709
Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH et al (2016) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. The Lancet 388(10062):2873–2884
Gegg ME, Cooper JM, Schapira AH, Taanman JW (2009) Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 4:e4756
Giacobini E, Gold G (2013) Alzheimer disease therapy—moving from amyloid-β to tau. Nat Rev Neurol 9:677–686
Gill SS, Patel NK, Hotton GR et al (2003) Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 9:589–595
Gitler AD, Dhillon P, Shorter J (2017) Neurodegenerative disease: models, mechanisms, and a new hope. Dis Models Mech 10:499–502
Gitto R, De-Luca L, Ferro S et al (2008) Computational studies to discover a new NR2B/NMDA receptor antagonist and evaluation of pharmacological profile. Chem Med Chem 3(10):1539–1548
Gitto R, De-Luca L, Ferro S et al (2014) Synthesis, modeling and biological characterization of 3-substituted-1H-indoles as ligands of GluN2B-containing Nmethyl- d-aspartate receptors. Bioorg Med Chem 22:1040–1048
Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L (2013) γ-Secretase inhibitors and modulators. Biochim Biophys Acta BBA-Biomembr 1828:2898–2907
Golding MC, Long CR, Carmell MA, Hannon GJ, Westhusin ME (2006) Suppression of prion protein in live-stock by RNA interference. Proc Natl Acad Sci 103:5285–5290
Golpich M, Rahmani B, Ibrahim NMAA et al (2015) Preconditioning as a potential strategy for the prevention of Parkinson’s disease. Mol Neurobiol 51:313–330
Gong CX, Iqbal K (2008) Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 15:2321–2328
Gong CX, Shaikh S, Grundke-Iqbal I, Iqbal K (1996) Inhibition of protein phosphatase-2B calcineurin activity towards Alzheimer abnormally phosphorylated τ by neuroleptics. Brain Res 41:95–102
Gravitz L (2011) Drugs: a tangled web of targets. Nature 475(7355):S9–S11. https://doi.org/10.1038/475S9a
Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA et al (2009) Tarenflurbil Phase 3 Study Group: effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302:2557–2564
Guttman M, Boileau I, Warsh J, Saint-Cyr JA, Ginovart N, McCluskey T, Houle S, Wilson A, Mundo E, Rusjan P et al (2007) Brain serotonin transporter binding in nondepressed patients with Parkinson’s disease. Eur J Neurol 14(5):523–528
Haapaniemi TH, Ahonen A, Torniainen P, Sotaniemi KA, Myllyla VV (2001) [123I]beta-CIT SPECT demonstrates decreased brain dopamine and serotonin transporter levels in untreated parkinsonian patients. Mov Disord 16(1):124–130
Hallock P, Michael A, Thomas (2012) Integrating the Alzheimer’s disease proteome and transcriptome: a comprehensive network model of a complex disease. J Integr Biol 16:37–49
Hamaguchi T, Ono K, Yamada M (2010) Review: curcumin and Alzheimer’s disease. CNS Neurosci Ther 16:285–297
Hanger DP, Anderton BH, Noble W (2009) Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med 15:112–119
Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM (1995) Direct evidence of oxidative injury produced by the Alzheimer’s β-amyloid peptide 1-40 in cultured hippocampal neurons. Exp Neurol 131:193–202
Hart FU, Bracher A, Hayer-Hart M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475:324–332
Hashimoto M, Rockenstei E, Mante M, Crews L, Bar-On P, Gage FH et al (2004) An anti-aggregation gene therapy strategy for lewy body disease utilizing β-synuclein lentivirus in a transgenic model. Gene Ther 11:1713–1723
Hastings TG (2009) The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J Bioenerg Biomembr 41(6):469–472
Haugarvoll K, Rademakers R, Kachergus JM et al (2008) LRRK2 R1441C Parkinsonism is clinically similar to sporadic Parkinson disease. Neurology 70:1456–1460
He DY, McGough NN, Ravindranathan A et al (2005) Glial cell line-derived neurotrophic factor mediates the desirable actions of the anti-addiction drug ibogaine against alcohol consumption. J Neurosci 25:619–628
Healy DG, Falchi M, O’Sullivan SS et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 7:583–590
Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. JAMA Neurol 60(8):1119–1122
Hedstrom KL, Murtie JC, Albers K, Calcutt NA, Corfas G (2014) Treating small fiber neuropathy by topical application of a small molecule modulator of ligand-induced GFRα/RET receptor signalling. Proc Natl Acad Sci 111(6):2325–2330
Heemels MT (2016) Neurodegenerative diseases. Nature 539(7628):179
Hershko A, Ciechanover A (1998) The ubiquitin system. Ann Rev Biochem 67:425–479
Herskovits AZ, Guarente L (2013) Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res 23:746–758
Hibiki K, Giovanni M (2017) Proteinopathies and OXP HOS dysfunction in neurodegenerative diseases. J Cell Biol 216:3917–3929
Holtcamp W (2012) The emerging science of BMAA: do cyanobacteria contribute to neurodegenerative disease? Environ Health Perspect 120(3):A110–A116
Hopkins CR (2011) ACS chemical neuroscience molecule spotlight on ELND006: another γ-secretase inhibitor fails in the clinic. ACS Chem Neurosci 2:279–280
Hou Y, Li S, Wu M et al (2016) Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am J Physiol Renal Physiol 310:F547–F559
Hsieh YL, Kan HW, Chiang H, Lee YC, Hsieh ST (2018) Distinct TrkA and Ret modulated negative and positive neuropathic behaviors in a mouse model of resiniferatoxin-induced small fiber neuropathy. Exp Neurol 300:87–99
Hu H, Li M (2016) Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem Biophys Res Commun 478(1):174–180
Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD et al (2006) Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 9:1520–1525
Hu S, Maiti P, Ma Q, Zuo X, Jones MR, Cole GM, Frautschy SA (2015) Clinical development of curcumin in neurodegenerative disease. Expert Rev Neurother 15:629–637
Hung SY, Fu WM (2017) Drug candidates in clinical trials for Alzheimer’s disease. J Biomed Sci 24:47
Huot P, Sgambato-Faure V, Fox SH, McCreary AC (2017) Serotonergic approaches in Parkinson’s disease: translational perspectives, an update. ACS Chem Neurosci 8(5):973–986
Infante J, Prieto C, Sierra M et al (2016) Comparative blood transcriptome analysis in idiopathic and LRRK2 G2019S-associated Parkinson’s disease. Neurobiol Aging 38(214):e1–e5
Iqbal K, Alonso ADC, Chen S, Chohan MO, El-Akkad E, Gong CX et al (2005) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta BBA Mol Basis Dis 1739:198–210
Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW (2003) Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62:389–397
Jacobsen JS, Reinhart P, Pangalos MN (2005) Current concepts in therapeutic strategies targeting cognitive decline and disease modification in Alzheimer’s disease. NeuroRx 2:612–626
Jacobsen H, Ozmen L, Caruso A, Narquizian R, Hilpert H, Jacobsen B, Terwel D, Tanghe A, Bohrmann B (2014) Combined treatment with a BACE inhibitor and anti- Aβ antibody gantenerumab enhances amyloid reduction in APPLondon mice. J Neurosci 34:11621–11630
Jana S, Sinha M, Chanda D et al (2011) Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim Biophys Acta 1812:663–673
Jenner P, Olanow CW (1996) Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 47:161S–170S
Johnson GVW, Hartigan JA (1999) Tau protein in normal and Alzheimer’s disease brain: an update. J Alzheimer’s Dis 1:329–351
Johnson BN, Bergera AK, Cortesea GP, LaVoie MJ (2012) The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. PNAS 109(16):6283–6288
Johri A, Beal MF (2012) Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 342:619–630
Kadowaki H, Nishitoh H, Urano F et al (2005a) Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ 12(1):19–24
Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K et al (2005b) Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ 12:19–24
Kahle PJ, Haass C (2004) How does parkin ligate ubiquitin to Parkinson’s disease? EMBO Rep 5:681–685
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386:896–912
Kanfi Y, Naiman S, Amir G et al (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483:218–221
Kastanenka KV, Bussiere T, Shakerdge N, Qian F, Weinreb PH, Rhodes K, Bacskai BJ (2016) Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J Neurosci 36:12549–12558
Keshet GI, Ovadia H, Taraboulos A, Gabizon R (1999) Scrapie-infected mice and PrP knockout mice share abnormal localization and activity of neuronal nitric oxide synthase. J Neurochem 72:1224–1231
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 287:19440–19451
Khan MT, Orhan I, Senol FS et al (2009) Cholinesterase inhibitory activities of some flavonoid derivatives and chosen xanthone and their molecular docking studies. Chem Biol Interact 181:383–389
Kiaei M (2008) Peroxisome proliferator-activated receptor gamma in amyotrophic lateral sclerosis and Huntington’s disease. PPAR Res 8:1–8
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S (1998) Mutations in the parkin gene cause autosomal recessive juvenile Parkinsonism. Nature 392:605–608
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ (2004) Hsp70 reduces α-synuclein aggregation and toxicity. J Biol Chem 279:25497–25502
Kokubo H, Kayed R, Glabe CG, Staufenbiel M, Saido TC, Iwata N et al (2009) Amyloid beta annular protofibrils in cell processes and synapses accumulate with aging and Alzheimer-associated genetic modification. Int J Alzheimer’s Dis 2009:689285. https://doi.org/10.4061/2009/689285
Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K et al (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci 874:1561–1565
Koren SA, Hamm MJ, Meier SE, Weiss BE, Nation GK, Chishti EA et al (2019) Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. https://doi.org/10.1007/s00401-019-01970-9
Krueger BA, Weil T, Schneider G (2009) Comparative virtual screening and novelty detection for NMDA-GlycineB antagonists. J Comput Aided Mol Des 23(12):869
Kumar A, Nisha CM, Silakari C et al (2016) Current and novel therapeutic molecules and targets in Alzheimer’s disease. J Formos Med Assoc 115(1):3–10
Kuzuhara S (2010) Treatment strategy of Alzheimer disease: pause of clinical trials of Aβ vaccine and next steps. Brain Nerve 62:659–666
Lang AE (2010) Clinical trials of disease modifying therapies for neurodegenerative diseases: the challenges and the future. Nat Med 1611:1223–1226
Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38–48
Lazarou M, Sliter DA, LA Kane JL et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314
Leong SL, Cappai R, Barnham KJ, Pham CL (2009) Modulation of alpha-synuclein aggregation by dopamine: a review. Neurochem Res 34(10):1838–1846
Leucht C, Vana K, Renner-Muller I et al (2004) Knock-down of the 37-kDa laminin receptor in mouse brain by transgenic expression of specific antisense LRP RNA. Transgenic Res 13:81–85
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR (2008) Physiology of the prion protein. Physiol Rev 88:673–728
Lipton SA (2005) The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res 2:155–165
Liraz O, Boehm-Cagan A, Michaelson D (2013) ApoE4 induces Aβ42, tau and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol Neurodegener 8:16
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22:1942–1950
Liu C, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9(2):106–118
Liu AK, Chang RC, Pearce RK, Gentleman SM (2015) Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer’s and Parkinson’s disease. Acta Neuropathol 129(4):527–540
Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, Basun H, Lannfelt L (2016) Safety and tolerability of BAN2401— a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther 8:14
Lozano AM, Lang AE, Hutchison WD, Dostrovsky JO (1998) New developments in understanding the etiology of Parkinson’s disease and in its treatment. Curr Opin Neurobiol 8:783–790
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 155:853–862
Luk KC, Mills IP, Trojanowski JQ, Lee VM (2008) Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry 47:12614–12625
Mackenzie G, Will R (2017) Creutzfeldt-Jakob disease: recent developments. F1000 Res 6:2053
MacLeod R, Hillert EK, Cameron RT, Baillie GS (2015) The role and therapeutic targeting of α, β and γ secretase in Alzheimer’s disease. Future Sci. https://doi.org/10.4155/fso159
Maiti P, Gregg LC, McDonald MP (2016) MPTP-induced executive dysfunction is associated with altered prefrontal serotonergic function. Behav Brain Res 298(Pt B):192–201
Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP (2010) Mitochondria targeted antioxidants protect against amyloid-b toxicity in Alzheimer’s disease neurons. J Alzheimer’s Dis 20:609–631
Manuelidis L (2013) Infectious particles, stress, and induced prion amyloids: a unifying perspective. Virulence 4:373–383
Marcade M, Bourdin J, Loiseau N, Peillon H, Rayer A, Drouin D et al (2008) Etazolate, a neuroprotective drug linking GABAA receptor pharmacology to amyloid precursor protein processing. J Neurochem 106:392–404
Marina SG, Oleg SG (2013) The molecular chaperone GRP78/BiP as a therapeutic target for neurodegenerative disorders: a mini review. J Genet Syndr Gene Ther 4(2):128
Marina SG, Arseniy S, Weijun C, Craig M, Layla FS, Max S, Jonathan HL, Alfred SL, Nicholas M, Oleg SG (2012) Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol Ther 20:1327–1337
Masliah E, Rockenstein E, Mante M et al (2011) Passive immunization reduces behavioural and neuropathological deficits in an alpha-synuclein transgenic model of lewy body disease. PLoS ONE 6:e19338
Mbazima V, Da-Costa-Dias B, Omar A, Jovanovic K, Weiss SF (2010) Interactions between PrP (c) and other ligands with the 37-kDa/67-kDa laminin receptor. Front Biosci 3667:1150–1163
McBride HM (2008) Parkin mitochondria in the autophagosome. J Cell Biol 183:757–759
McLean PJ, Kawamata H, Shariff S et al (2002a) Torsin-A and heat shock proteins act as molecular chaperones: suppression of α-synuclein aggregation. J Neurochem 83:846–854
McLean S, Naish R, Reed L, Urry S, Vicenzino B (2002b) A pilot study of the manual force levels required to produce manipulation induced hypoalgesia. Clin Biomech 17:304–308
Mehta M, Adem A, Sabbagh M (2012) New acetylcholinesterase inhibitors for Alzheimer’s disease. Int J Alzheimer’s Dis. https://doi.org/10.1155/2012/728983
Melancon BJ, Tarr JC, Panarese JD, Wood MR, Lindsley CW (2013) Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Dis Today 18(23–24):1185–1199
Min SW, Sohn PD, Cho SH, Swanson RA, Gan L (2013) Sirtuins in neurodegenerative diseases: an update on potential mechanisms. Front Aging Neurosci 5:1–9
Mojtaba G, Elham A, Zahurin M, Raymond AA, Norlinah MI, Abolhassan A (2017) Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis treatment CNS. Neurosci Therapeut 23:5–22
Mondragón-Rodríguez S, Perry G, Zhu X, Boehm J (2012) Amyloid beta and tau proteins as therapeutic targets for Alzheimer’s disease treatment: rethinking the current strategy. Int J Alzheimer’s Dis. https://doi.org/10.1155/2012/630182
Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR (2009) An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J Alzheimers Dis 16:741–761
Moreira PI, Zhu X, Wang X, Lee HG, Nunomura A, Petersen RB, Perry G, Smith MA (2010) Mitochondria: a therapeutic target in neurodegeneration. Biochim Biophys Acta 1802:212–220
Morimoto RI (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22:427–1438
Muchowski PJ, Wacker JL (2005a) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22
Muchowski PJ, Wacker JL (2005b) Modulation of neurodegeneration by molecular chaperones. Neuroscience 6(1):11
Mudher A, Lovestone S (2002) Alzheimer’s disease do tauists and baptists finally shake hands? Trends Neurosci 25:22–26
Murray ED, Buttner EA, Price BH (2012) Depression and psychosis in neurological practice. In: Daroff R, Fenichel G, Jankovic J, Mazziotta J (eds) Bradley’s neurology in clinical practice, 6th edn. Elsevier/Saunders, Philadelphia
Narendra D, Tanaka A, Suen DF et al (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Niforou K, Cheimonidou C, Trougakos IP (2014) Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol 2:323–332
Nikolaev A (2009) APP binds DR6 to cause axon pruning and neuron death via distinct caspases. Nat 457:981–989
Onyango IG, Lu JH, Rodova M, Lezi E, Crafter AB, Swerdlow RH (2010) Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta 1802:228–234
Osswald G (2018) BioArctic announces positive topline results of BAN2401 Phase 2b at 18 months in early Alzheimer’s disease. BioArctic Press Release, Stockholm
Outeiro TF, Kontopoulos E, Altmann SM et al (2007) Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317:516–519
Ovadia H, Rosenmann H, Shezen E, Halimi M, Ofran I, Gabizon R (1996) Effect of scrapie infection on the activity of neuronal nitricoxide synthase in brain and neuroblastoma cells. J Biol Chem 271:16856–16861
Pajares M, Jiménez-Moreno N, Dias IH et al (2015) Redox control of protein degradation. Redox Biol 6:409–420
Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W (2009) Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 164:541–551
Panegyres PK, Armari E (2013) Therapies for human prion diseases. Am J Neurodegen Dis 2:176–186
Panza F, Solfrizzi V, Seripa D, Imbimbo BP, Lozupone M, Santamato A, Zecca C, Barulli MR, Bellomo A, Pilotto A, Daniele A (2016) Tau-centric targets and drugs in clinical development for the treatment of Alzheimer’s disease. Biomed Res Int 2016:3245935
Parson CG, Danysz W, Dekundy A, Pulte I (2013) Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox Res 24:358–369
Peden AH, Head MW, Diane LR, Jeanne EB, James WI (2004) Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364(9433):527–529
Peoc’h K, Manivet P, Beaudry P et al (2000) Identification of three novel mutations E196 K, V203I, E211Q in the prion protein gene PRNP in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Human Mutat 15(5):482
Petersen RB, Siedlak SL, Lee HG et al (2005) Redox metals and oxidative abnormalities in human prion diseases. Acta Neuropathol 110:232–238
Petri S, Kiaei M, Damiano M et al (2006) Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem 98:1141–1148
Pfeifer A, Eigenbrod S, Al-Khadra SM et al (2006) Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-Infected mice. J Clin Invest 116:3204–3210
Pflanz H, Vana K, Mitteregger G et al (2009) Microinjection of lentiviral vectors expressing small interfering RNAs directed against laminin receptor precursor mRNA prolongs the pre-clinical phase in scrapie-infected mice. J Gen Virol 90:269–274
Pickhardt M, Gazova Z, Von-Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow EM et al (2005) Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J Biol Chem 280:3628–3635
Pickrell AM, Youle RJ (2015) The roles of PINK1 parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85(2):257–273
Pietri M, Dakowski C, Hannaoui S, Alleaume-Butaux A, Hernandez-Rapp J, Ragagnin A et al (2013) PDK1 decreases TACE-mediated [alpha]-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat Med 19:1124–1131
Pinna A (2014) Adenosine A2A receptor antagonists in Parkinson’s disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 28:455–474
Polymeropoulos MH, Lavedan C, Leroy E et al (1997) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Protter D, Lang C, Cooper AA (2012) α-Synuclein and mitochondrial dysfunction: a pathogenic partnership in Parkinson’s disease. Parkinson’s Dis 2012:829207. https://doi.org/10.1155/2012/829207
Prusiner SB, Scott MR, DeArmond SJ, Cohen FE (1998) Prion protein biology. Cell 93:337–348
Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS (2002) Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem 83(4):973–983
Qin B, Cartier L, Dubois-dauphin M, Li B, Serrander L, Krause KH (2006) A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging 27(11):1577–1587
Rankin CA, Sun Q, Gamblin TC (2005) Pseudo-phosphorylation of tau at Ser202 and Thr205 affects tau filament formation. Mol Brain Res 138:84–93
Rascol O, Brooks DJ, Melamed E et al (2005) LARGO Study Group Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations LARGO, Lasting effect in Adjunct therapy with Rasagiline given once daily, study: a randomised, double-blind, parallel-group trial. Lancet 365:947–954
Reimann RR, Sonati T, Hornemann S et al (2016) Differential toxicity of antibodies to the prion protein. PLoS Pathog 28:e1005401
Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE et al (2010) 11 C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 9:363–372
Rogers MB (2018) To block tau’s proteopathic spread, antibody must attack its mid-region. Alzforum. https://www.alzforum.org/news/conference-coverage/blocktaus-proteopathic-spread-antibody-ustattack-its-mid-region
Roy KK, Tota S, Tripathi T, Chander S, Nath C, Saxena AK (2012) Lead optimization studies towards the discovery of novel carbamates as potent AChE inhibitors for the potential treatment of Alzheimer’s disease. Bioorg Med Chem 20:6313–6320
Rubenstein R (2017) Possible causes of Alzheimer’s disease related Amyloid-β plaques and neurofibrillary tangles. Sci J Lander Coll Arts Sci 10(2):3
Sabbagh MN (2009) Drug development for Alzheimer’s disease: where are we now and where are we headed? Am J Geriatric Pharmacother 7:167–185
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M et al (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. New Engl J Med 370:322–333
Samii A, Nutt JG, Ransom BR (2004) Parkinson’s Dis. Lancet 363:1183–1193
Sanchez MG, Morissette M, Di Paolo T (2013) Estradiol and brain serotonin reuptake transporter in long-term ovariectomized parkinsonian monkeys. Prog Neuro-Psychopharmacol Biol Psychiatry 45:170–177
Sattar H (2013) Fundamentals of pathology. Pathoma LLC; 2nd edition 2013
Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88:611–638
Scarpulla RC (2011) Metabolic control of mitochondrial Biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813:1269–1278
Scott NK (2013) Infection prevention: review and update for neurodiagnostic technologists. Neurodiag J 534:271–288
Sharma J, Johnston MV, Hossain MA (2014) Sex differences in mitochondrial biogenesis determine neuronal death and survival in response to oxygen glucose deprivation and reoxygenation. BMC Neurosci 15:1–14
Shelat PB, Chalimoniuk M, Wang JH et al (2008) Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem 106(1):45–55
Sheng R, Lin X, Zhang J, Chol KS, Huang W, Yang B et al (2009) Design, synthesis and evaluation of flavonoid derivatives as potent AChE inhibitors. Bioorg Med Chem 17:6692–6698
Shimura H, Hattori N, Kubo SI et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25:302–305
Shin-ichiro K, Taku H, Masashi T, Nobutaka H (2013) Can parkin be a target for future treatment of Parkinson’s disease? Expert Opin Ther Targets 17:10
Sidorova YA, Saarma M (2016) Glial cell line-derived neurotrophic factor family ligands and their therapeutic potential. Mol Biol 50(4):521–531
Sidorova YA, Bespalov MM, Wong AWTO et al (2017) A novel small molecule GDNF receptor RET agonist, BT13, promotes neurite growth from sensory neurons in vitro and attenuates experimental neuropathy in the rat. Front Pharmacol 8:365
Simonovitch S, Schmukler E, Pinkas-Kramarski R et al (2016) Impaired autophagy in APOE4 astrocytes. J Alzheimer’s Dis JAD 51(3):915–927
Slevin JT, Gerhardt GA, Smith CD, Gash DM, Kryscio R, Young B (2005) Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg 102:216–222
Smith DG, Cappa R, Barnham KJ (2007) The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim Biophys Acta 1768(8):1976–1990
Snead D, Eliezer D (2014) Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol 23:292–313
Sofic E, Riederer P, Heinsen H et al (1988) Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm 74:199–205
Sorensen EB, Conner SD (2010) γ-secretase dependent cleavage initiates notch signalling from the plasma membrane. Trafficking 11:1234–1245
Sveinbjornsdottir S (2016) The clinical symptoms of Parkinson’s disease. J Neurochem 139:318–324
Szeto HH (2008) Cell-permeable, mitochondrial-targeted, peptide antioxidants.Drug addiction. Springer, New York, pp 535–546
Tagliavini F, Pilleri G (1983) Basal nucleus of Meynert, a neuropathological study in Alzheimer’s disease, simple senile dementia, Pick’s disease and Huntington’s chorea. J Neurol Sci 62(1–3):243–260
Tamgüney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P (2008) Genes contributing to prion pathogenesis. J Gen Virol 89:1777–1788
Tan Z, Shi L, Schreiber SS (2009a) Differential expression of redox factor-1 associated with beta-amyloid-mediated neurotoxicity. Open Neurosci J 3:26–34
Tan Z, Shi L, Schreiber SS (2009b) Differential expression of redox factor-1 associated with beta-amyloid-mediated neurotoxicity. Open Neurosci J3:26–34
Tan SK, Hartung H, Sharp T, Temel Y (2011) Serotonin-dependent depression in Parkinson’s disease: a role for the subthalamic nucleus? Neuropharmacol 61(3):387–399
Tariot PN, Lopera F, Langbaum JB et al (2018) The alzheimer’s prevention initiative autosomal- dominant alzheimer’s disease trial: a study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal- dominant Alzheimer’s disease, including a placebo- treated noncarrier cohort. Alzheimers Dement 4:150–160
Taymansa JM, Greggio E (2016) LRRK2 Kinase Inhibition as a therapeutic strategy for Parkinson’s disease, where do we stand. Curr Neuropharmacol 14:214–225
Tilly G, Chapuis J, Vilette D, Laude H, Vilotte JL (2003) Efficient and specific downregulation of prion protein expression by RNAi. Biochem Biophys Res Commun 305:548–551
Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (2004) The importance of neuritic plaques and tangles to the development and evolution of AD. Neurol 62:1984–1989
Tokugawa K, Yamamoto K, Nishiguchi M et al (2003) XIB4035, a novel nonpeptidyl small molecule agonist for GFRα-1. Neurochem Int 42(1):81–86
Ultsch M, Li B, Maurer T, Mathieu M, Adolfsson O, Muhs A, Pfeifer A, Pihlgren M, Bainbridge TW, Reichelt M, Ernst JA (2016) Structure of crenezumab complex with Aβ shows loss of β- hairpin. Sci Rep 6:39374
Urwin PJ, Mackenzie JM, Llewelyn CA, Will RG, Hewitt PE (2016) Creutzfeldt-Jakob disease and blood transfusion: updated results of the UK transfusion medicine epidemiology review study. Vox Sang 110(4):310–316
Valente EM, Abou-Sleiman PM, Caputo V et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Van-Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, Ittner LM (2010) Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci 107:13888–13893
Varadaraju KR, Kumar JR, Mallesha L et al (2013) Virtual screening and biological evaluation of piperazine derivatives as human acetylcholinesterase inhibitors. Int J Alzheimers Dis. https://doi.org/10.1155/2013/653962
Vella LJ, Cappai R (2012) Identification of a novel amyloid precursor protein processing pathway that generates secreted N-terminal fragments. FASEB J 267:2930–2940
Vellas B, Sol O, Snyder PJ, Ousset PJ, Haddad R, Maurin M et al (2011) EHT0202 in Alzheimer’s disease: a 3-month, randomized, placebo-controlled, double-blind study. Curr Alzheimer Res 8:203–212
Wang X, Su BO, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29(28):9090–9103
Wang A, Das P, Switzer RC, Golde TE, Jankowsky JL (2011) Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-β immunotherapy. J Neurosci 31:4124–4136
Ward HJ, Everington D, Cousens SN, Smith-Bathgate B, Leitch M, Cooper S, Heath C, Knight RS, Smith PG, Will RG (2006) Risk factors for variant Creutzfeldt-Jakob disease: a case-control study. Ann Neurol 59(1):111–120
West T (2016) Safety, tolerability and pharmacokinetics of ABBV-8E12, a humanized anti-tau monoclonal antibody, in a Phase I, single ascending dose, placebo controlled study in subjects with progressive supranuclear palsy. J Prev Alzheimers Dis 3:285
White MD, Mallucci GR (2009) Therapy for prion diseases Insights from the use of RNA interference. Prion 3:121–128
Will RG (2003) Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull 66:255–265
Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, Mackenzie J, Estibeiro K, Green AJ, Knight RS (2000) Diagnosis of new variant Creutzfeldt–Jakob disease. Ann Neurol 47(5):575–582
Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, Maguire RP, Blennow K, Lundmark J, Staufenbiel M, Orgogozo JM (2012) Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double- blind, placebo- controlled first- in-human study. Lancet Neurol 11:597–604
Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR (1996) Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA 93:11213–11218
Wolfe MS (2012) γ-Secretase inhibitors and modulators for Alzheimer’s disease. J Neurochem 1(120):89–98
Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H et al (2005) Beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme BACE1 and gamma-secretase. J Biol Chem 280:23009–23017
Wu Z, Huang X, Feng Y et al (2006) Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1 alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci 103:14379–14384
Xiang W, Menges S, Schlachetzki JC et al (2015) Posttranslational modification and mutation of histidine 50 trigger alpha synuclein aggregation and toxicity. Mol Neurodegener 10:8
Yanamandra K, Kfoury N, Jiang H et al (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80:402–414
Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2015) Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2:278–288
Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF (2009) Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal 11:2095–2104
Yin W, Signore AP, Iwai M, Cao GD, Gao YQ, Chen J (2008) Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 39:3057–3063
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12(1):9–14
Zhang H, Bosch-Marce M, Shimoda LA et al (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283:10892–10903
Zhao J, Nussinov R, Ma B (2017) Mechanisms of recognition of amyloid- β (Aβ) monomer, oligomer, and fibril by homologous antibodies. J Biol Chem 292:18325–18343
Zhou Y, Danbolt NC (2014) Glutamate as a neurotransmitter in the healthy brain. J Neural Transm 121:799–817
Zourlidou A, Payne-Smith MD, Latchman DS (2004) HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem 88:1439–144810
Funding
This study was not supported by any funding agency.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that there are no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dar, K.B., Bhat, A.H., Amin, S. et al. Elucidating Critical Proteinopathic Mechanisms and Potential Drug Targets in Neurodegeneration. Cell Mol Neurobiol 40, 313–345 (2020). https://doi.org/10.1007/s10571-019-00741-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-019-00741-0