
Abstract
Oxidative glutamate toxicity plays a vital role in the neurodegeneration diseases, including Alzheimer’s diseases (AD). This study set out with the aim to investigate the beneficial effects of fangchinoline (FAN), a natural alkaloid, against glutamate-induced oxidative damage, and to clarify the underlying cellular and biochemical mechanisms. FAN prevented HT22 cells death from oxidative glutamate cytotoxicity in a dose-dependent manner, and significantly attenuated the overproduction of intracellular reactive oxygen species (ROS) and reversed the reduction of superoxide dismutase (SOD) activity induced by glutamate. Further investigations on the underlying mechanisms demonstrated that FAN potently up-regulated the protein level of nuclear factor erythroid 2-related factor 2 (Nrf2) and heme oxygenase (HO-1), in glutamate-exposed HT22 cells. The protective effects of FAN were almost completely antagonized by inhibitor of Nrf2. Subsequent studies revealed that FAN could down-regulate Kelch-like ECH-associated protein 1 (Keap1) in both mRNA level and protein level. To sum up, our result demonstrated the protective effects of FAN against glutamate-induced oxidative neuronal damage, and for the first time clarified the anti-oxidative mechanisms of FAN involve activating endogenous antioxidant defense system including enhancing SOD activity and regulating Keap1/Nrf-2 antioxidation signaling through modulation of Keap1 expression. Above results shed more light on the molecular mechanisms of FAN’s neuroprotective effects, and may provide important clues for the drug development in preventing oxidative stress-associated neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glutamate, a main excitatory neurotransmitter in central nervous system, plays a vital role in cell metabolism and physiology (Coyle and Puttfarcken 1993). However, excessive glutamate will suppress the uptake of cysteine by system X −C , the glutamate/cysteine antiporter, causing the inhibition of synthesis of glutathione (GSH) and accumulation of ROS (Albrecht et al. 2010; Murphy et al. 1989). This non-receptor-mediated glutamate-induced neurotoxicity is called oxidative glutamate toxicity, which is known as one of the most prominent pathological changes in neurodegenerative diseases, including AD (Coyle and Puttfarcken 1993; Tan et al. 2001). Consequently, agents that can suppress oxidative glutamate cytotoxicity are expected to be beneficial in the therapy of these neurodegenerative diseases.
With the fact that current active free radical scavengers exhibit unsatisfied potency to counteract oxidative glutamate toxicity, alternatively, increased efforts have been put on searching for novel agents to stimulate endogenous antioxidant defense systems. Recently, Nrf2, an indispensable part in the endogenous neuroprotective process (Jiang et al. 2017; Motohashi and Yamamoto 2004), was proved to play a crucial role in oxidative glutamate neurotoxicity (Morroni et al. 2018; Prasansuklab et al. 2017). Under physiological conditions, cytoplasmic Nrf2 maintains at very low concentrations in the intracellular level through Keap1-mediated ubiquitylation (Itoh et al. 1999; Kobayashi et al. 2004). In the case of oxidative insult, the detachment of Nrf2 and Keap1 leads to the nucleus translocation of Nrf2 and forms heterodimers with one of the small Maf protein (Velichkova and Hasson 2005). Consequently, the heterodimers recognize the antioxidant responsive element (ARE) and regulate the expression of HO-1 (Pae et al. 2008). Recently, activation of Keap1-Nrf2/ARE signaling pathway has been proposed to be a potential therapeutic approach against oxidative glutamate toxicity (Kim et al. 2017; Prasansuklab and Tencomnao 2018; Wei et al. 2018), as well as oxidative stress-associated multiple neurodegeneration diseases (Dodson et al. 2018; Morroni et al. 2018; Sandberg et al. 2014).
Natural compounds, known for diverse structures, are the main sources of drug discovery (Harvey and Cree 2010). In the present study, our natural compounds library was screened to discover novel candidates that can effectively protect neuronal cells against glutamate-induced oxidative damage, employing a widely used mouse hippocampal HT22 cell line to mimic excessive glutamate-induced endogenous oxidative stress for its lack of functional ionotropic glutamate receptor (Fukui et al. 2010; Murphy et al. 1989; Tan et al. 1998). To our delight, fangchinoline (FAN), an bis-benzylisoquinoline alkaloid firstly extracted from plants of Stephania tetrandra, was found to robustly protected neuronal cells against glutamate-induced injury (Fan et al. 2017a). Although studies have shown that FAN possesses diverse bioactivities (Choi et al. 2000; Merarchi et al. 2018), whether FAN can protect neuronal cells from oxidative glutamate toxicity has not been reported yet. This study investigates the protective effects of FAN against oxidative glutamate neurotoxicity and elucidates the involvement of Keap1-Nrf2 antioxidant defense system in the pharmacological effects of FAN.
Materials and Methods
HT22 Cell Culture
HT22 cell line (Jennio Biotechnology Co., Ltd., Guangzhou, China) was maintained in DMEM medium (Life Tech, Grand Island, USA) containing 10% FBS (Gibco, Grand Island, USA) with addition of antibiotics, and cultured at 37 °C under 5% CO2.
Drug Treatment
FAN purchased from Selleck (Shanghai, China) was dissolved using dimethyl sulfoxide (DMSO) and diluted with DMEM medium. HT22 neuronal cells were seeded in 96-well plates and six-well plates (5 × 104 cells per mL) (Corning, Corning, NY, USA). Twelve hours after seeding, cells were treated with indicated concentrations of FAN for 2 h and then incubated with glutamate (Sigma-Aldrich, St. Louis, MO, USA) for another 24 h. In a separate experiment, Brusatol (80 nM), an inhibitor of Nrf2, was added to cells for 2 h before FAN treatment.
Cell Viability Assay
Cell morphology was observed under the microscope (Nikon, TE200, USA), and cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltertrazolium bromide (MTT, Sangon Biotech, Shanghai, China) assay. Cells were incubated with 0.5 mg/mL MTT (100 µL per well) for 3 h at 37 °C. The cultured medium was then replaced by 100% DMSO (100 μL per well) and gently shaking for 5 min. The absorbance was recorded at a wavelength of 490 nm (DTX 800, Beckman Coulter, USA).
Measurement of ROS Level
HT22 neuronal cells were treated with FAN for 2 h before glutamate stimulation. Then replace the DMEM medium to 100 μL Na+ medium (132 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1.4 mM MgCl2·6H2O, 1.2 mM NaH2PO4·2H2O, 6 mM Glucose, 10 mM HEPES, pH 7.4) containing 10 μM H2-DCFDA. Cells were cultured at 37 °C for 45 min followed by three times washes with Na+ medium and photographed under the fluorescence microscope (TE200, Nikon, Melville, USA). Then cells were extracted using 100 μL 1% SDS buffer (1% SDS and 5 mM Tris–HCl) and fluorescence was recorded at Ex/Em = 485/520 nm (DTX 800, Beckman Coulter, USA).
Intracellular SOD Activity Measurement
HT22 neuronal cells were treated with FAN for 2 h before 6 h glutamate stimulation. The SOD enzymatic activity was determined using a commercial assay kit (Jiancheng Biochemical, Nanjing, China). Pierce BCA protein assay kit (Thermo Scientific, IL, USA) was used to measure the amount of protein.
Free Radical Scavenging Assay
The direct radical scavenging ability was determined by 1,1-diphenyl-2-picrylhydrazyl (DPPH, Alfa Aesar, China) assay. FAN (10 μL) in different concentrations were incubated with 150 μM DPPH ethanol solution (90 μL) for 30 min at room temperature in dark, finally measured at 517 nm wavelength (EnVision, PerkinElmer, Waltham, MA, USA). The DPPH scavenge rate (%) = [1 − (Asample − Ablank)/(Acontrol − Ablank)] × 100%.
Real-Time Polymerase Chain Reaction (RT-PCR)
Total cellular RNA was extracted using Trizol reagent (Life Tech, Grand Island, USA). PrimeScript RT Master Mix kit and SYBR Premix ExTaq kit (Takara Biomedical Technology, Beijing, China) were used for reverse-transcribed and RT-PCR according to the manufacturer’s instruction. Primers are as follows: Keap1: forward, 5′-CCAGATTGACAGCGTGGTTC-3′; reverse, 5′-GGTTGAAGAACTCCTCCTGCT-3′; GAPDH: forward, 5′-GGTCCCAGCTTAGGTTCATCA-3′; reverse, 5′-CCGTTCACACCGACCTTCA-3′.
Transient Transfection with siRNA
HT22 neuronal cells were seeded for 12 h at 2 × 104 cells per mL in 96-well plates and six-wells plates. Cells were transfected with 10 nM siRNA duplex (Jima Biotechnology, Shanghai, China) using Lipofectamine RNAiMAX (Thermo Scientific, Rockford, IL, USA). After 24 h treatment with siRNA, the medium was replaced and the cells were treated with FAN for 2 h and then incubated with 10 mM glutamate for 24 h. The sequences used to knockdown mouse Keap1 expression were as follows: forward, 5′-GAAGCAAAUUGAUCAACAAT-3′; reverse, 5′-UUGUUGAUCAAUUUGCUUCTT-3′.
Western Blotting Analysis
Western blotting analysis was performed as previous described (Feng et al. 2019). The antibodies we used were as follows: anti-HO-1 (1:5000, Sigma-Aldrich, St. Louis, MO, USA), anti-β-actin (1:10000, Sigma-Aldrich, St. Louis, MO, USA), anti-Nrf2 (1:500, Abcam, Cambridge, UK), anti-Keap1 (1:5000, Proteintech, Wuhan, China), and horseradish peroxidase-conjugated secondary antibodies (1:5000, Kangchen Biotechnology, Shanghai, China).
Statistical Analysis
Data in this study are expressed as the mean ± standard error of mean (SEM). One-way ANOVA with Turkey’s test was used for the multiple comparisons, and unpaired student’s t test was used to analyze the differences between two groups. P-value below 0.05 was considered significant.
Results
FAN Attenuates Glutamate-Induced Neurotoxicity
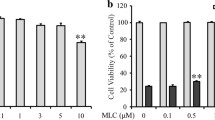
In order to identify the effect of FAN on glutamate-induced neurotoxicity, HT22 cells were treated with FAN for 2 h before glutamate stimulation. Stimulation of glutamate alone on HT22 cells showed changes in cell morphology which became shrinkage and debris, while pretreatment with 5 μM FAN remarkably attenuated these morphological damages (Fig. 1a). Consistent with the cell morphological changes, significant reduction of cell viability induced by glutamate could also be observed. In contrast, pretreatment with FAN (1, 3, and 5 μM) dose-dependently alleviated glutamate-induced cell viability reduction (Fig. 1b). FAN at 5 μM showed a maximum protection, and 5 μM was therefore chosen as the optimal concentration for further studies. Effects of FAN alone on cell viability was also investigated, and result showed various concentration of FAN had no statistical different effect on the cell viability of HT22 cells (Fig. 1c).

FAN protected HT22 cells against glutamate-induced cytotoxicity. a Phase-contrast micrographs (10 ×) of cells subjected to different treatments. Scale bar = 100 μm. b Cells were pretreated with indicated concentrations of FAN for 2 h, and then exposed to 10 mM glutamate for 24 h. c HT22 cells were treated with indicated concentration of FAN alone for 24 h. Cell viability was determined using the MTT assay. Data were from three independent experiments. ***P < 0.001 versus the control group, ###P < 0.001 versus the glutamate-treated group
FAN Alleviates the Overproduction of ROS and the Reduction of SOD Induced by Glutamate
We identified the antioxidant properties of FAN in both cellular and molecular level. In HT22 cells, intracellular ROS level was markedly increased after glutamate stimulation for 6 h, while the generation of ROS was markedly alleviated when cells were pretreated with 5 μM FAN (Fig. 2a, b). To further evaluate the antioxidant activity of FAN in HT22 cells, SOD enzymatic activity was measured. Intracellular SOD enzymatic activity was considerably decreased with glutamate exposure in 6 h; however, this trend can be reversed by pretreatment with FAN for 2 h (Fig. 2c). DPPH scavenging assay, an approach widely used for the evaluation of compounds’ antioxidant property (Brand-Williams et al. 1995), was used to measure the direct radical scavenging effect of FAN, with trolox as a positive control. The result showed that FAN had a stronger DPPH scavenging capacity (EC50 = 26.70 ± 1.37 μM) compared to that of Trolox (EC50 = 37.05 ± 0.60 μM) (Fig. 2d).

FAN inhibited glutamate-induced ROS formation and restored the depletion of intracellular SOD enzymatic activity. Cells were pretreated with 5 μM FAN for 2 h and then exposed to 10 mM glutamate for 6 h. a H2DCF-DA fluorescence were analyzed by visual observation of cell morphology through fluorescence microscopy (10 ×), scale bar = 100 μm (A). b The production of ROS was measured by assessing the changes in the DCFH-DA fluorescence intensity. c The SOD activity was normalized as a ratio relative to the control activity. d Direct radical scavenging capacity of FAN was determined using DPPH scavenging activity assay. Data were from four independent experiments. *P < 0.05 versus the control group, #P < 0.05 versus the glutamate-treated group
FAN Increases HO-1 Protein Level
Nrf2/ARE, an important endogenous defense mechanism, is considered as an expected therapeutic target against oxidative glutamate toxicity, and HO-1 is one of the most essential downstream effectors of Nrf2/ARE signaling pathway (Lavrovsky et al. 2000). Therefore, the influence of FAN on the protein level of HO-1 was firstly evaluated. Results showed that within 6 h, treatment with 5 μM of FAN alone time-dependently up-regulated the protein level of HO-1, with significant changes at 3 and 6 h treatment (Fig. 3a). Glutamate exposure also induced a significant enhancement in HO-1 level, while pretreatment with FAN 2 h before glutamate stimulation persistently up-regulated the protein level of HO-1 (Fig. 3b).

FAN increased HO-1 expressions in HT22 cells. a Western blot analysis of HO-1 protein level after treatment with FAN for indicated time period. b Western blot analysis of HO-1 protein level in different treatment groups. Data were from four independent experiments. *P < 0.05, ***P < 0.001 versus the control group, ##P < 0.01 versus the glutamate-treated group
FAN Increases Nrf2 Protein Level in HT22 Cells
Whether the enhancement of HO-1 in protein level related to the activation of its upstream protein, Nrf2, needed to be further investigated. And in attempt to figure out this hypothesis, we investigated the protein level of Nrf2. Treatment with 5 μM FAN dramatically up-regulated the protein level of Nrf2 within 6 h compared to the control group (Fig. 4a and Supplementary Fig. 1). Similar to the changes in HO-1 level, glutamate exposure led to a significant enhancement in the level of Nrf2, and pretreatment with FAN 2 h before glutamate stimulation persistently up-regulated the protein level of Nrf2 in the whole cell lysate (Fig. 4a). As generally known, in the normal condition, Nrf2 is presented in the cytoplasm and translocated into the nucleus under oxidative stress. To further investigate whether FAN treatment alters the Nrf2 nucleus translocation, we isolated nuclear and cytoplasmic extracts and found that 6-h treatment with FAN up-regulated the protein level of nuclear Nrf2, indicating that Nrf2 was translocated to the nucleus (Fig. 4b). We then hypothesized that the role of FAN in prevention glutamate-induced cell death might be relevant to the activation of Nrf2. In attempt to further probe the role of Nrf2 in the protective effects of FAN, brusatol, an inhibitor of Nrf2 which can provoke a rapid depletion of Nrf2 protein (Olayanju et al. 2015) was used. Compared with FAN-treated group, co-incubation with 5 μM FAN and 80 nM brusatol potently suppressed the enhancement of Nrf2 protein and concomitantly reduced the up-regulation of HO-1 stimulated by FAN (Fig. 4c). In addition, co-incubation with FAN and brusatol blocked the neuroprotective effects of FAN (Fig. 4d).

Nrf2 mediates the protection of FAN against glutamate-induced cytotoxicity in HT22 cells. a Western blot analysis of Nrf2 protein level in the whole cell lysate after treatment with FAN for 2 h and exposed to glutamate for 6 h. b Western blot analysis of Nrf2 protein level in nucleus and cytoplasm. c Cells were pretreated with/without 80 nM brusatol for 2 h, followed by incubating with/without 5 μM FAN for 6 h. Western blot analysis of Nrf2 and HO-1 protein levels after treatment. d Cells were pretreated with/without 80 nM brusatol for 2 h, followed by incubating with/without 5 μM FAN for 2 h and then exposed to glutamate for 24 h. Cell viability was measured by MTT. Data were from four independent experiments. ***P < 0.001 versus the control group, #P < 0.05, ###P < 0.001 versus the glutamate-treated group, &P < 0.05 versus the FAN-treated group, $$$P < 0.001 versus FAN in combination with glutamate-treated group
FAN Decreases the Expression of Keap1
Under physiological condition, Nrf2 binds to Keap1 as a complex and undergoes ubiquitination and proteasomal degradation in cytoplasm (Nguyen et al. 2009). In contrast, Keap1 was degraded and separated form Nrf2 for the modification of critical cysteine residues, triggering the nuclear translocation of Nrf2 under oxidative insult (Bryan et al. 2013). To investigate whether regulation of Keap1 mediated the protective effects of FAN, the expression of Keap1 was measured. Within 6 h, treatment with FAN alone time-dependently down-regulated the protein level of Keap1 (Fig. 5a). Similarly, treatment with FAN alone also decreased the expression of Keap1 in mRNA level significantly in 6 h (Fig. 5b). Moreover, we overexpressed Keap1 to investigate the influence on FAN-induced changes in Nrf2 and HO-1 levels. The result showed that, FAN alone could up-regulate the protein levels of Nrf2 and HO-1, while in the presence of Keap1 overexpression, these changes were significantly reversed (Supplementary Fig. 2). To further probe the role of Keap1 in the protective effects of FAN, we used small interfering RNA to knockdown Keap1. Transfection with Keap1 siRNA significantly reduced the expression of Keap1 in protein level compared to nontargeting control (NC) siRNA (Supplementary Fig. 3). Furthermore, no significant changes were shown in cell viability between siKeap1-FAN-glutamate-treated group and siKeap1-glutamate-treated group, and silencing Keap1 could prevent cells against oxidative glutamate toxicity (Fig. 5c).

FAN decreases the protein and mRNA levels of Keap1. a Western blot analysis of Keap1 protein level after HT22 cells were treated with FAN for indicated times. b RT-PCR results of Keap1 mRNA level after HT22 cells were treated with FAN for indicated times. c Cells were transfected with siRNA for 48 h and cell viability was measured by MTT after different treatments. Data were from four independent experiments. *P < 0.05, **P < 0.01 versus the control group, ##P < 0.01 versus the glutamate-treated group
Discussion
The primarily oxidative stress triggered by excessive glutamate, which called oxidative glutamate toxicity, has been considered to contribute largely to neuronal injury in many neurodegenerative diseases (Coyle and Puttfarcken 1993; Tan et al. 2001). And here, we firstly discovered that FAN, a natural alkaloid, potently activates the endogenous survival pathway Keap1-Nrf2 signaling pathway to protect HT22 cells against oxidative glutamate neurotoxicity.
The present study identified that FAN markedly prevented HT22 cells from oxidative glutamate toxicity (Fig. 1a, b). Generally, glutamate-induced neurotoxicity has been shown in two pathways, one is the ionotropic glutamate receptor-mediated excitotoxicity, and the second distinct pathway is non-receptor-mediated oxidative toxicity (Murphy et al. 1989). Although previous studies have reported that FAN could suppress the release of glutamate and inhibit ionotropic glutamate receptor-medicated excitotoxicity by the inhibition of Ca2+ influx (Kim et al. 2001; Koh et al. 2003; Lin et al. 2009), which are quite different from our findings on protective effects of FAN against glutamate-induced but non-receptor-medicated oxidative neuronal damage, as present study was performed under excessive glutamate exposure and in a cellular system (HT22 cells) lack of the functional ionotropic glutamate receptor. Besides, FAN (0.3 to 5 μM) had no significant influence on the cell viability when treated alone (Fig. 1c), suggesting the neuroprotective effects of FAN were not based on the simple effect of cell proliferation.
The accumulation of ROS and reduction of SOD activity are two primary pathological processes in glutamate-induced neuronal cell death (Kang et al. 2014; Chao et al. 2014) and an variety of molecules has been reported to prevent neuron death from oxidative glutamate cytotoxicity by blocking ROS production and SOD deduction (Jeong et al. 2014; Li et al. 2017). We thus measured the influence of FAN on the intracellular ROS level and SOD activity. Similar as previous study (Chao et al. 2014), our results indicated that high concentration glutamate could consequently lead to the overproduction of ROS and reduce the enzymatic activity of SOD, while FAN treatment significantly reduced the generation of glutamate-induced intracellular ROS and reversed the reduction of SOD activity (Fig. 2b, c), indicating the neuroprotective effects of FAN may happened in a ROS and SOD-dependent manner. Although previous study has shown that FAN inhibited hydrogen peroxide-induced generation of ROS (Koh et al. 2003), in contrast to H2O2-induced cytotoxicity model, in oxidative glutamate toxicity model, ROS are generated endogenously rather than the supplementary from externally that leading to cell injury, suggesting that FAN may potently active the endogenous antioxidant defense system besides direct ROS scavenging. In fact, our results found that FAN had the capability to scavenge DPPH free radicals (Fig. 2d), which is in compliance with the previous research (Gulcin et al. 2010). As the DPPH scavenging ratio of FAN was only 14.3% at 5 μM (Fig. 2d), the working concentration against oxidative glutamate toxicity, indicating that the direct DPPH scavenging capacity of FAN may only partially contribute to its prevention effects against oxidative glutamate cytotoxicity.
Taken together, we hypothesized that other mechanisms may be involved in FAN against neurotoxicity, such as the activation of endogenous antioxidant signaling pathway. We further assessed the association between FAN’s protection against glutamate oxidative damage and regulation of Keap1-Nrf2, one of the main endogenous antioxidant signaling pathways to resist oxidative stress (Fan et al. 2017b). To our delight, we observed that FAN significantly up-regulated the protein level of HO-1 (Fig. 3) and Nrf2 (Fig. 4a, b) in HT22 cells, indicating that FAN may activate Nrf2/ARE pathway. Our subsequent study employing brusatol, a unique inhibitor of Nrf2, demonstrated that inhibiting the Nrf2 signaling leads to a reduction of HO-1 in protein level (Fig. 4c), and blockage of the protective effects of FAN against oxidative glutamate neurotoxicity (Fig. 4d), indicating that the protective role of FAN against oxidative glutamate neurotoxicity might be largely mediated by activing Nrf2.
Nrf2 is a main factor of the Keap1-Nrf2-HO-1 signaling pathway. Upon activation, Nrf2 is separated from Keap1 and translocated into the nucleus and consequently enhance nuclear Nrf2 level (Prasad 2016; Xiang et al. 2018). And we wonder whether the up-regulation of Nrf2 protein level by FAN was related to the regulation of Keap1. Our study found that FAN treatment decreased the expressions of Keap1 in both mRNA and protein levels (Fig. 5a, b). When silencing Keap1, no significant difference was found in the cell viability between glutamate-exposed groups with or without FAN treatment (Fig. 5c), indicating that the oxidative damage induced by glutamate can be largely alleviated through downregulation of Keap1 and FAN didn’t exhibit additional benefits when inhibiting Keap1 signaling, which suggests that the protection of FAN against glutamate-induced oxidative cytotoxicity may closely related to its regulation on Keap1 expression. Our study indicates that FAN may directly down-regulate Keap1 gene expression and consequently promote the activation of Nrf2-dependent anti-oxidative pathway, which is mechanistically distinct from most active compounds which activating Nrf2 signaling through inhibiting Keap1 ubiquitination (Lee and Jeong 2016; Prasansuklab et al. 2017; Wei et al. 2018). Our results together with the beneficial effects of Keap1 silencing against glutamate-induced cell damage suggest that the direct regulation of Keap1 expression may be an alternative and effectively approach to battle oxidative glutamate toxicity.
In summary, our work demonstrates that FAN prevents HT22 cell death from glutamate-induced non-receptor-medicated oxidative neuronal damage and for the first time reveals the underlying mechanism may involve regulating Keap1/Nrf-2 antioxidation signaling via downregulation of Keap1 expression, which may provide new clues for the anti-oxidative drug development. Further investigation will be needed to judge this approach and the systematically evaluate the efficacies and mechanisms of FAN.
References
Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A (2010) Mechanisms of oxidative glutamate toxicity the glutamate cystine antiporter system xc-as a neuroprotective drug target. CNS Neurol Disord: Drug Targets 9:373–382. https://doi.org/10.2174/187152710791292567
Brand-Williams W, Cuvelier ME, Berset C (1995) Use of a free-radical method to evaluate antioxidant activity. Food Sci Technol-Leb 28:25–30. https://doi.org/10.1016/S0023-6438(95)80008-5
Bryan HK, Olayanju A, Goldring CE, Park BK (2013) The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 85:705–717. https://doi.org/10.1016/j.bcp.2012.11.016
Chao XJ, Chen ZW, Liu AM, He XX, Wang SG, Wang YT, Liu PQ, Ramassamy C, Mak SH, Cui W, Kong AN, Yu ZL, Han YF, Pi RB (2014) Effect of tacrine-3-caffeic acid, a novel multifunctional anti-Alzheimer’s dimer, against oxidative-stress-induced cell death in HT22 hippocampal neurons: involvement of Nrf2/HO-1 pathway. CNS Neurosci Ther 20:840–850. https://doi.org/10.1111/cns.12286
Choi HS, Kim HS, Min KR, Kim Y, Lim HK, Chang YK, Chung MW (2000) Anti-inflammatory effects of fangchinoline and tetrandrine. J Ethnopharmacol 69:173–179. https://doi.org/10.1016/S0378-8741(99)00141-5
Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695. https://doi.org/10.1126/science.7901908
Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD (2018) Modulating NRF2 in disease: timing is everything. Annu Rev Pharmacol Toxicol 59:555–575. https://doi.org/10.1146/annurev-pharmtox-010818-021856
Fan B, Zhang X, Ma Y, Zhang A (2017a) Fangchinoline induces apoptosis, autophagy and energetic impairment in bladder cancer. Cell Physiol Biochem 43:1003–1011. https://doi.org/10.1159/000481698
Fan H, Shu Q, Guan X, Zhao J, Yan J, Li X, Liu J, Jia Z, Shi J, Li J (2017b) Sinomenine protects PC12 neuronal cells against H2O2-induced cytotoxicity and oxidative stress via a ROS-dependent up-regulation of endogenous antioxidant system. Cell Mol Neurobiol 37:1387–1398. https://doi.org/10.1007/s10571-017-0469-1
Feng HX, Li CP, Shu SJ, Liu H, Zhang HY (2019) A11, a novel diaryl acylhydrazone derivative, exerts neuroprotection against ischemic injury in vitro and in vivo. Acta Pharmacol Sin 40:160–169. https://doi.org/10.1038/s41401-018-0028-4
Fukui M, Choi HJ, Zhu BT (2010) Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic Biol Med 49:800–813. https://doi.org/10.1016/j.freeradbiomed.2010.06.002
Gulcin I, Elias R, Gepdiremen A, Chea A, Topal F (2010) Antioxidant activity of bisbenzylisoquinoline alkaloids from Stephania rotunda: cepharanthine and fangchinoline. J Enzyme Inhib Med Chem 25:44–53. https://doi.org/10.3109/14756360902932792
Harvey AL, Cree IA (2010) High-throughput screening of natural products for cancer therapy. Planta Med 76:1080–1086. https://doi.org/10.1055/s-0030-1250162
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86. https://doi.org/10.1101/gad.13.1.76
Jeong EJ, Hwang L, Lee M, Lee KY, Ahn MJ, Sung SH (2014) Neuroprotective biflavonoids of Chamaecyparis obtusa leaves against glutamate-induced oxidative stress in HT22 hippocampal cells. Food Chem Toxicol 64:397–402. https://doi.org/10.1016/j.fct.2013.12.003
Jiang S, Deng C, Lv J, Fan C, Hu W, Di S, Yan X, Ma Z, Liang Z, Yang Y (2017) Nrf2 weaves an elaborate network of neuroprotection against stroke. Mol Neurobiol 54:1440–1455. https://doi.org/10.1007/s12035-016-9707-7
Kang Y, Tiziani S, Park G, Kaul M, Paternostro G (2014) Cellular protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun 5:3672. https://doi.org/10.1038/ncomms4672
Kim SD, Oh SK, Kim HS, Seong YH (2001) Inhibitory effect of fangchinoline on excitatory amino acids-induced neurotoxicity in cultured rat cerebellar granule cells. Arch Pharm Res 24:164–170. https://doi.org/10.1007/Bf02976485
Kim NT, Lee DS, Chowdhury A, Lee H, Cha BY, Woo JT, Woo ER, Jang JH (2017) Acerogenin C from Acer nikoense exhibits a neuroprotective effect in mouse hippocampal HT22 cell lines through the upregulation of Nrf-2/HO-1 signaling pathways. Mol Med Rep 16:1537–1543. https://doi.org/10.3892/mmr.2017.6682
Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24:7130–7139. https://doi.org/10.1128/MCB.24.16.7130-7139.2004
Koh SB, Ban JY, Lee BY, Seong YH (2003) Protective effects of fangchinoline and tetrandrine on hydrogen peroxide-induced oxidative neuronal cell damage in cultured rat cerebellar granule cells. Planta Med 69:506–512. https://doi.org/10.1055/s-2003-40647
Lavrovsky Y, Chatterjee B, Clark RA, Roy AK (2000) Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp Gerontol 35:521–532. https://doi.org/10.1016/s0531-5565(00)00118-2
Lee DS, Jeong GS (2016) Butein provides neuroprotective and anti-neuroinflammatory effects through Nrf2/ARE-dependent haem oxygenase 1 expression by activating the PI3K/Akt pathway. Br J Pharmacol 173:2894–2909. https://doi.org/10.1111/bph.13569
Li Z, Chen X, Lu W, Zhang S, Guan X, Li Z, Wang D (2017) Anti-oxidative stress activity is essential for Amanita caesarea mediated neuroprotection on glutamate-induced apoptotic HT22 cells and an Alzheimer’s disease mouse model. Int J Mol Sci 18:1623. https://doi.org/10.3390/ijms18081623
Lin TY, Lu CW, Tien LT, Chuang SH, Wang YR, Chang WH, Wang SJ (2009) Fangchinoline inhibits glutamate release from rat cerebral cortex nerve terminals (synaptosomes). Neurochem Int 54:506–512. https://doi.org/10.1016/j.neuint.2009.02.001
Merarchi M, Sethi G, Fan L, Mishra S, Arfuso F, Ahn KS (2018) Molecular targets modulated by fangchinoline in tumor cells and preclinical models. Molecules (Basel, Switzerland) 23:2538. https://doi.org/10.3390/molecules23102538
Morroni F, Sita G, Graziosi A, Turrini E, Fimognari C, Tarozzi A, Hrelia P (2018) Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of Alzheimer’s disease involves Nrf2/HO-1 pathway. Aging Dis 9:605–622. https://doi.org/10.14336/AD.2017.0903
Motohashi H, Yamamoto M (2004) Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 10:549–557. https://doi.org/10.1016/j.molmed.2004.09.003
Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT (1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2:1547–1558. https://doi.org/10.1016/0896-6273(89)90043-3
Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284:13291–13295. https://doi.org/10.1074/jbc.R900010200
Olayanju A, Copple IM, Bryan HK, Edge GT, Sison RL, Wong MW, Lai ZQ, Lin ZX, Dunn K, Sanderson CM, Alghanem AF, Cross MJ, Ellis EC, Ingelman-Sundberg M, Malik HZ, Kitteringham NR, Goldring CE, Park BK (2015) Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med 78:202–212. https://doi.org/10.1016/j.freeradbiomed.2014.11.003
Pae HO, Kim EC, Chung HT (2008) Integrative survival response evoked by heme oxygenase-1 and heme metabolites. J Clin Biochem Nutr 42:197–203. https://doi.org/10.3164/jcbn.2008029
Prasad KN (2016) Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech Ageing Dev 153:41–47. https://doi.org/10.1016/j.mad.2016.01.002
Prasansuklab A, Tencomnao T (2018) Acanthus ebracteatus leaf extract provides neuronal cell protection against oxidative stress injury induced by glutamate. BMC Complement Altern Med 18:278. https://doi.org/10.1186/s12906-018-2340-4
Prasansuklab A, Meemon K, Sobhon P, Tencomnao T (2017) Ethanolic extract of Streblus asper leaves protects against glutamate-induced toxicity in HT22 hippocampal neuronal cells and extends lifespan of Caenorhabditis elegans. BMC Complement Altern Med 17:551. https://doi.org/10.1186/s12906-017-2050-3
Sandberg M, Patil J, D’Angelo B, Weber SG, Mallard C (2014) NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 79:298–306. https://doi.org/10.1016/j.neuropharm.2013.11.004
Tan SL, Wood M, Maher P (1998) Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J Neurochem 71:95–105. https://doi.org/10.1046/j.1471-4159.1998.71010095.x
Tan S, Schubert D, Maher P (2001) Oxytosis: a novel form of programmed cell death. Curr Top Med Chem 1:497–506. https://doi.org/10.2174/1568026013394741
Velichkova M, Hasson T (2005) Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol 25:4501–4513. https://doi.org/10.1128/MCB.25.11.4501-4513.2005
Wei Y, Liu D, Zheng Y, Hao C, Li H, Ouyang W (2018) Neuroprotective effects of kinetin against glutamate-induced oxidative cytotoxicity in HT22 cells: involvement of Nrf2 and heme oxygenase-1. Neurotox Res 33:725–737. https://doi.org/10.1007/s12640-017-9811-0
Xiang Y, Ye W, Huang C, Yu D, Chen H, Deng T, Zhang F, Lou B, Zhang J, Shi K, Chen B, Zhou M (2018) Brusatol enhances the chemotherapy efficacy of gemcitabine in pancreatic cancer via the Nrf2 signalling pathway. Oxid Med Cell Longev 2018:2360427. https://doi.org/10.1155/2018/2360427
Acknowledgements
We gratefully acknowledge financial support of this work by Grant Nos. 81872859, 81703507, 81522045 from the National Natural Science Foundation of China.
Author information
Authors and Affiliations
Contributions
FB, LT, and HZ designed the study; FB performed all the study; FB and LT analyzed pharmacological data; FB, LT, and HZ wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bao, F., Tao, L. & Zhang, H. Neuroprotective Effect of Natural Alkaloid Fangchinoline Against Oxidative Glutamate Toxicity: Involvement of Keap1-Nrf2 Axis Regulation. Cell Mol Neurobiol 39, 1177–1186 (2019). https://doi.org/10.1007/s10571-019-00711-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-019-00711-6