
Abstract
Sinomenine (SN), a purified alkaloid from Chinese herb Sinomenium acutum that was used preferentially in the treatment of rheumatoid diseases, has exerted neuroprotective effects and anti-inflammatory properties in many previous studies. Some studies have revealed that the antioxidant property of SN, acting mainly through inhibiting NADPH oxidase activation, was involved in the beneficial effects of SN. However, SN belongs to the family of dextrorotatory morphinan analogues, which may initiate elevation of reactive oxygen species (ROS) levels. Thus in the present report, we conducted studies to examine its impact and mechanism on the resistance of PC12 neuronal cells to oxidative stress. Precondition with SN (0.1–5 μM) for 12 h significantly decreased H2O2-induced cytotoxicity and remarkably alleviated oxidative injury. However, SN exhibited little direct free radical scavenging property in vitro and induced “appropriate” production of ROS in PC12 cell. Interestingly, the SN-triggering ROS production served as a signal to activate the Nrf2 antioxidant system including Nrf2, HO-1, and NQO-1, which was inhibited by the antioxidant trolox. Furthermore, Nrf2 knockdown largely attenuated the beneficial effects of SN precondition on oxidative stress. In conclusion, our findings suggested that SN increased the resistance to oxidative stress in neuronal cells via a ROS-dependent up-regulation of endogenous antioxidant system, and this mechanism may be involved in the neuroprotection of SN.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sinomenine (SN), a purified alkaloid from Chinese herb Sinomenium acutum, has been used for the clinical treatment of inflammatory diseases for many centuries in China (Xu et al. 2008). Previous studies have demonstrated that the pharmacological profiles of SN include immunosuppression, anti-inflammation, protection against acute lung injury, anti-apoptotic properties, and so on (Bao et al. 2005; Feng et al. 2006; Li et al. 2013; Qian et al. 2007). In recent 5 years, some studies have revealed that the SN provided significant neuroprotection against neurological diseases, including brain ischemia (Wu et al. 2011) and Parkinson’s disease (PD) (Qian et al. 2007). Although mechanistic studies raised some potential targets for SN, much less is known about the molecular mechanism by which SN exhibits neuroprotective effects.
Reactive oxygen species (ROS), including superoxide (O2−•), hydrogen peroxide (H2O2), and hydroxyl radical (OH•), can function either as signaling molecules or as cellular toxicants that involved in a variety of physiological and pathological processes (Carvalho et al. 2016; Kim et al. 2015; Lin and Beal 2006; Popa-Wagner et al. 2013). It is well known that morphine generates cellular ROS in a concentration- and time-dependent manner and ROS is proverbially involved in the effect of morphine (Ma et al. 2015). Based on its molecular structure, SN belongs to the family of dextrorotatory morphinan analogues (as shown in Fig. 1a) and therefore may also trigger the production of ROS (Jin et al. 2008). However, some reports have indicated that SN exhibited the antioxidant properties mainly through inhibiting NADPH oxidase (NOX), which contributed to its protective effects (Qian et al. 2007). Up to now, very little is known about the mechanism underlying SN-mediated antioxidation. A recent report has indicated that SN activated nuclear factor erythroid-derived 2-like 2 (Nrf2) signaling, a key antioxidant mechanism maintains intracellular redox homeostasis underlying oxidative stress (Qin et al. 2016a; b). Given the reported antioxidant properties of SN, here, we conducted studies to examine its impact and mechanism on the resistance of PC12 cells to oxidative stress. Interestingly, it was observed that SN protected PC12 neuronal cells against H2O2-induced cytotoxicity through increased resistance to oxidative stress which is mediated by a ROS-dependent up-regulation of endogenous antioxidant system, and thus might act as a novel neuroprotector for neurologic diseases associated with oxidative stress.

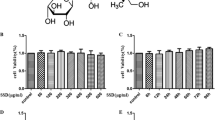
SN protected PC12 cells against H2O2-induced cytotoxicity. a Chemical structure of SN. b PC 12 cells were incubated with different concentrations of H2O2 (25, 50, 100, and 200 μM) for 12 h, the H2O2-induced cell toxicity was evaluated by MTT assay. c PC12 cells were incubated with indicated concentrations of SN (0.1, 0.5, 1, 5, 10, and 50 μM) for 24 h. The effects of SN on cell viability were evaluated by MTT assay. d PC12 cells were incubated with 50 μM SN for 24, 48, or 72 h. The effects of SN on cell viability were evaluated by MTT assay. e–f PC12 cells (e) and primary cultured cortical neurons (f) were pre-incubated 24 h with or without SN (0.1, 0.5, 1, and 5 μM) prior to H2O2 (200 μM) exposure for 12 h. Protective effects of SN on H2O2-induced cell toxicity were evaluated by MTT assay. g PC12 cells were co-incubated with H2O2 (200 μM) and SN (0.1, 0.5, 1, and 5 μM) for 12 h. Protective effects of SN on H2O2-induced cell toxicity were evaluated by MTT assay. Resveratrol (Res) was used as a positive control. Results were expressed as a relative change in comparison with controls, which was set to 100%. Data are expressed as mean ± SEM. *p < 0.05 or ***p < 0.001 versus control group, # p < 0.05, ## p < 0.01 or ### p < 0.001 versus H2O2 treatment group
Materials and Methods
Materials
SN and resveratrol were obtained from the National Institutes for Food and Drug Control (Beijing, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), heat-inactivated horse serum, streptomycin, penicillin, glutamine, B27 supplements, and trizol reagent were acquired from Gibco/Invitrogen Inc. (Carlsbad, CA, USA). 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide (MTT) and peroxide hydrogen (H2O2) were purchased from Sigma-Aldrich Chemical Inc. (St. Louis, MO, USA). The ApoGSH™Glutathione (GSH) Colorimetric Detection Kit was obtained from BioVision Inc. (Milpitas, CA, USA). Malondialdehyde (MDA) assay kit and total superoxide dismutase (SOD) assay kit were acquired from Beyotime Biotechnology (Shanghai, China). The lucigenin-enhanced chemiluminescence with a colorimetric assay kit was obtained from GENMED Scientifics Inc. (Shanghai, China). The RevertAid-TM First Strand cDNA Synthesis system was purchased from Transgene Inc. (Beijing, China). SYBR Green I PCR Premix was obtained from TaKaRa Biotechnology (Dalian, China). The anti-Nrf2 and anti-HO-1 polyclonal antibodies were purchased from Cell signaling Technology (Beverly, USA). The anti-NQO-1 polyclonal antibody and anti-NOX2 monoclonal antibody were purchased from Abcam (Cambridge, UK). The anti-β-actin and appropriate peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). All other chemicals were of analytical grade.
Cell Culture and Experimental Treatment
The PC12 cell line was acquired from Cell Bank of Chinese Academic of Science (Shanghai, China). PC12 cells were grown in DMEM supplemented with 5% FBS, 10% heat-inactivated horse serum, 100 U/ml streptomycin, and 100 μg/ml penicillin. Cultures were kept at 37 °C in a humidified atmosphere incubator with 5% CO2/95% air. The culture medium was replaced every two days. All experiments were carried out 24 h after cells were seeded.
The isolation and culture of the cortical neurons were slightly modified from our previous studies (Wu et al. 2011). All experimental procedures were performed in accordance with guidelines for animal experiments established by Henan University of Science and Technology. Neonatal Sprague–Dawley (SD) rats (day 0–3) were obtained from the Experimental Animal Center of Henan University of Science and Technology. Briefly, cortex of neonatal SD rats was carefully isolated and dissected, and then tissue was incubated with 0.125% trypsin in PBS for 20 min at 37 °C. After collected by centrifugation at 800 × g for 5 min, cells were resuspended in DMEM/F-12 (1:1) with 10% FBS, 5 U/ml penicillin, 5 g/ml streptomycin, and 0.5 mM glutamine. Finally, cells were plated onto poly-d-lysine coated 96-well (for the measurements of cell viability) or 6-well plates (for the measurements of gene expression) and kept at 37 °C in a 5% CO2 incubator. After 24 h, the culture medium was changed to DMEM medium supplemented with 2% B27, and the cortical neurons were fed with fresh medium twice weekly. Glial cells were minimized by treating the culture with cytarabine (10 M) on day 3, and experiments were performed on day 7–9.
SN was freshly prepared as stock solution in dimethyl sulfoxide (DMSO) and diluted with phosphate-buffered saline (PBS) before the experiment. The final concentration of DMSO was <0.05%. No detectable effect of DMSO was found in the experiments. To produce oxidative stress, H2O2 was freshly prepared from 30% stock solution prior to each experiment. PC12 cells or primary cultured cortical neurons were pre-incubated with various concentrations of SN for 1, 6, or 24 h before exposure to H2O2 (200 μM). Assays for cell viability, MDA, GSH, SOD, NOX, quantitative analysis of gene expression, and western blotting were performed at different time periods after H2O2 was added.
Assay of Cell Viability
The cytoprotective activity of SN on H2O2-induced cell injury was evaluated by MTT reduction assay. PC12 cells or primary cultured cortical neurons were placed into a 96-well plate for 24 h and then pretreated with vehicle alone or indicated concentrations of SN for 24 h. After the pretreatment, the culture was added to H2O2 (200 μM) for another 12 h and the MTT solution (dissolved in PBS) was added to the medium with the final concentration of 0.5 mg/ml. Four h later, cells were lysed in DMSO and the amount of MTT formazan was assessed by determining the absorbance at 570 nm using a microplate reader (SpectraMax 250, Molecular Devices Inc., Sunnyvale, CA). Survival of the control groups was defined as 100%, and cell viability in the treated groups was expressed as a percentage of the control groups.
Electron Spin Resonance (ESR) Measurement
The ESR measurement was conducted as previously described (Fan et al. 2015). N-tert-butyl-α-phenylnitrone (PBN) was used as a free radical trapper and ESR signals were detected with a Bruker e-scan ESR spectrometer (Bruker, Karlsruhe, Germany). As a standard of the reactant of OH· with PBN, we produced hydroxyl radical (OH·) by the Fenton reaction in the mixture of H2O2 (0.5 mM) and FeSO4 (0.2 mM) in the presence of PBN (30 mM). The spin traps, SN (0.1, 0.5, 1, and 5 μM) were added before the Fe(II) and H2O2. Samples (20 μl) were loaded into a quartz tube and the ESR spectra were recorded at room temperature.
The ESR microwave power was set to 4.88 mW. The modulation frequency was 9.76 GHz. The time constant was 20.48 ms. The conversion time was 20.48 ms and a sweep time of 10.49 s was used. Each sample was scanned for a total of 20 times. A sweep width of 70 G was used for experiments with PBN. The receiver gain was set to 3.17 × 103. Simulation and fitting of the ESR spectra were performed using the Bruker WinEPR program.
Measurement of MDA
After the treatment, PC12 cells were lysed and then the cell lysate was centrifuged at 1600 × g for 10 min at 4 °C. The supernatants were removed to measure the levels of MDA according to the manufacturer’s instructions.
Measurement of GSH
The content of GSH was evaluated according to the manufacturer’s recommendations for the ApoGSH™ Glutathione Colorimetric Detection Kit. Briefly, cells (5 × 105 cells) were harvested by centrifugation at 700 × g for 5 min at 4 °C and then the supernatants were removed. The cell pellets were resuspended in 0.5 ml ice-cold PBS and lysed in 80 μl ice-cold glutathione buffer. Then the samples were mixed with 20 μl 5-sulfosalicylic acid (SSA) and centrifuged at 8000 × g for 10 min. The supernatants (20 μl) were added to the reaction mix (160 μl) at room temperature. After 10 min incubation, the substrate solution (20 μl) was added to the mixture for a further 10 min and the content of GSH was quantified via determining the absorbance at 415 nm using a microplate reader (SpectraMax250, Molecular Devices Inc., Sunnyvale, CA). The standard curve was obtained from the absorbance of the diluted GSH standard which was incubated in the mixture as in samples.
Measurement of SOD Activity
After the treatment, PC12 cells were lysed and then the cell lysate was centrifugated at 1600 × g for 10 min at 4 °C. The supernatants were removed to measure the activity of SOD according to the manufacturer’s instructions. Briefly, total activity of SOD was assessed by the scavenging of superoxide anion generated by xanthine, while the remained superoxide anion oxidizes WST-8 to WST-8 formazan. Therefore, the absorption of formed WST-8 formazan is negatively correlative to the total activity of SOD, which can be evaluated by determining the absorbance at 450 nm using a microplate reader (SpectraMax250, Molecular Devices Inc., Sunnyvale, CA).
Quantitative Analysis of Gene Expression
The procedure for quantitative real-time PCR (qPCR) was performed as described in our previous studies (Fan et al. 2015). Total cellular RNA was extracted from PC12 cells or primary cultured cortical neurons using trizol reagent following the procedure described by the manufacturer. The concentration and purity of RNA were determined spectrophotometrically at ratio 260/280, respectively. cDNA was synthesized from the total RNA using the RevertAid-TM First Strand cDNA Synthesis system. The Primers (Sangon, Shanghai, China) used for qPCR analysis of Nrf2, HO-1, NQO-1, and β-actin were as follows: 5′-CCTCGCTGGAAAAAGAAGTG-3′ and 5′-GGAGAGGATGCTGCTGAAAG-3′ for Nrf2; 5′-CAGGTGATGCTGACAGAGGA-3′ and 5′-TCTCTGCAGGGGCAGTATCT-3′ for HO-1; 5′-TTCTCTGGCCGATTCAGAGT-3′ and 5′-TCCAGACGTTTCTTCCATCC-3′ for NQO-1; and 5′-GCTCCTTCGTTGCCGGTCC-3′ and 5′-CTCTTGCTCTGGGCCTCGTCA-3′ for β-actin. qPCR reactions were performed with SYBR Green I PCR Premix using an ABI 7500 real-time PCR system (Applied Biosystems, Carlsbad, USA). For each target mRNA, 2 μl cDNA was used as a template in each 20 μl qPCR reactions. The protocol was 2 min at 95 °C, 40 cycles of 15 s at 95 °C, 15 s at 62 °C, and 30 s at 72 °C. Each assay was run at least in triplicate in separate experiment, and negative controls were generated by substituting cDNA with distilled water. The purity of qPCR products was assessed by melting curves, and the relative expression levels of target genes were determined by comparative CT method by normalizing to β-actin and relative to a calibrator (2−△△Ct).
Western Blotting
After treatments, PC12 cells were washed three times with PBS and lysed in ice-cold lysis buffer (1 mM EDTA, 1 mM phenylmethanesulfonyl fluoride, 3 mM Na3VO4, 20 mM NaF, 50 mM Tris–HCl, 100 mM NaCl, 1% Nonidet P-40 (v/v), 2.0 l g/ml aprotinin, 2.0 l g/ml leupeptin, and 2.0 l g/ml pepstatin A). Equal amounts of protein (20 μg) from above lysates were separated in 10–15% SDS-PAGE gels and transferred onto nitrocellulose membranes by using a transfer cell system (Bio-Rad, California, USA). The membranes were blocked with 5% bovine serum albumin (BSA) diluted in TBS/0.1% Tween-20 for 1 h at room temperature. After blocking, the membranes were incubated with the following primary antibodies (anti-Nrf2 at 1 : 1000 dilution, anti-HO-1 at 1 : 1000 dilution, anti-NQO-1 at 1 : 1000 dilution, anti-NOX2 at 1: 2000 dilution, and anti-β-actin at 1 : 2000 dilution) overnight at 4 °C. Then the membranes were washed three times with TBS/0.1% Tween-20 and appropriate secondary antibodies were used to detect the proteins. Immunoblots were developed on microchemi (DNR, Jerusalem, Israel) by using the enhanced chemiluminescence technique (ECL, Pierce, Rockford, USA). All assays were performed at least three times. An image analysis system and ImageJ 1.49 were used to determine the densitometry of immunoblots relative to the loading control.
Nrf2 Gene Silencing Experiments
siRNA oligonucleotides targeting Nrf2 was purchased from Santa Cruz Biotechnology. Cells were transfected with 25 nM of Nrf2 or scrambled-siRNA oligonucleotides using Lipofectamine RNAiMAX according to the manufacturer’s instructions (Invitrogen).
Measurement of NADPH Oxidase Activity (NOX Activity)
The activity of NOX was evaluated by lucigenin-enhanced chemiluminescence with a colorimetric assay kit following the manufacturer’s instructions. Briefly, the supernatant of cell lysates was incubated with oxidative cytochrome c in a quartz cuvette at 30 °C for 3 min, and then NADPH which served as the NOX substrate was added into the reaction mixture for another 15 min. The change of absorbance at 550 nm was recorded by a spectrophotometer and the activity of NOX was measured by calculating cytochrome c reduction in per minute. The relative NOX activity was expressed as a percentage of the control value.
Statistics
Data from experiments were analyzed with the statistical program SPSS 18.0 software (SPSS, Chicago, IL). Comparison between two groups was evaluated by an unpaired and two-sided Student’s t test. ANOVAs and post hoc tests (Fisher’s LSD) were used to analyze differences in different treatment groups. Data are presented as mean ± SEM. Differences between experimental conditions were considered statistically significant when p < 0.05.
Results
SN Pre-conditioning Protected PC12 Cells and Primary Cultured Neurons Against H2O2-Induced Cytotoxicity
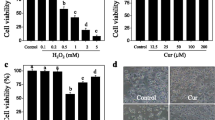
Initially, the cytotoxic potential of H2O2 was measured. As shown in Fig. 1b, the rate of cell viability was dose-dependently inhibited when PC12 cells were incubated with indicated concentrations of H2O2 (25, 50, 100, and 200 μM) for 12 h (n = 10, *p < 0.05 or ***p < 0.001 vs. control). When PC12 cells were exposed to different concentrations of SN (0.1, 0.5, 1, 5, 10 or 50 μM) alone for 24 h, no statistically significant difference in the viability of PC12 cells was observed (Fig. 1c). However, PC12 cells incubated with 50 μM SN for 72 h exhibited obvious toxic effects (n = 10, *p < 0.05 vs. control, Fig. 1d). To further evaluate the neuroprotective effects of SN, PC12 cells and primary cultured cortical neurons were pretreated with indicated concentrations of SN for 24 h and then exposed to H2O2 (200 μM). After 12 h, the H2O2-induced cell injury was determined by MTT assay. As shown in Fig. 1e–f, incubation with 200 μM H2O2 significantly decreased the rate of cell viability by 52.67 ± 5.60% (n = 10, ***p < 0.001 vs. control, Fig. 1e) in PC12 cells and 43.43 ± 6.38% (n = 10, ***p < 0.001 vs. control, Fig. 1f) in primary neurons. When cells were pretreated with SN (0.1, 0.5, 1, and 5 μM) for 24 h, H2O2-induced cell toxicity was efficiently attenuated in a dose-dependent manner. SN at a concentration of 5 μM rescued the H2O2-induced decrease in viability rate by about 92.95 ± 3.45% (n = 10, ### p < 0.001 vs. H2O2, Fig. 1e) in PC12 cells and 77.37 ± 7.59% (n = 10, ## p < 0.01 vs. H2O2, Fig. 1f) in primary neurons, respectively. Finally, the protective effects of SN were also assessed when PC12 cells were co-treated with H2O2 and SN, and similar but less effective results of SN were obtained. As shown in Fig. 1g, cotreatment with 5 μM SN rescued the H2O2-induced decrease in rate of cell viability by 78.43 ± 6.51% (n = 10, ## p < 0.01 vs. H2O2).
SN pre-conditioning remarkably alleviated H2O2-induced oxidative stress in PC12 cells
Oxidative stress is considered to play an essential role in H2O2-dependent cytotoxicity; therefore, the effect of SN on ROS was studied by evaluating the levels of malondialdehyde (MDA), the content of glutathione (GSH), and superoxide dismutase (SOD) activity.
MDA has been extensively studied as an indicator of lipid peroxidation which is one of the earliest recognized and most widely studied manifestations of oxygen toxicity in biology. As shown in Fig. 2a, a significant increase of MDA levels (2.09 ± 0.16 nm/mg pro) compared to control group (0.77 ± 0.06 nm/mg pro) was observed in PC12 cells exposed to H2O2 (n = 5, ***p < 0.001 vs. control). Pretreatment with SN at a concentration of 1 and 5 μM remarkably decreased the content of MDA by 1.57 ± 0.12 nm/mg pro (n = 5, # p < 0.05 vs. H2O2) and 1.28 ± 0.13 nm/mg pro (n = 5, ## p < 0.01 vs. H2O2), respectively. Furthermore, antioxidant agents, such as GSH, and antioxidant enzymes, such as SOD, are the primary defense systems to protect biological systems from oxidative stress. As shown in Fig. 2b, c, exposure of PC12 cells to H2O2 significantly reduced the content of GSH (6.44 ± 0.46 μm/mg pro) and the activity of SOD (31.68 ± 3.40 U/mg pro) to 3.40 ± 0.28 μm/mg pro and 12.03 ± 1.45 U/mg pro, respectively, as compared to control group (n = 5, **p < 0.01 vs. control). However, the decrease of GSH levels and SOD activity induced by H2O2 was dose-dependently attenuated when PC12 cells were pre-incubated with SN at a concentration of 1 and 5 μM for 24 h (n = 5, ## p < 0.01 or ### p < 0.001 vs. H2O2). These findings indicated that SN protected PC12 cells against H2O2-induced cell injury mainly via increasing resistance to oxidative stress.

SN remarkably alleviated H2O2-induced oxidative stress in PC12 cells. PC12 cells were pre-incubated 24 h with or without SN (1 and 5 μM) prior to H2O2 (200 μM) exposure for 12 h. The levels of MDA (a), the content of GSH (b), and the activity of SOD (c) were evaluated. Res was used as a positive control. Results were expressed as a relative change in comparison with controls, which was set to 100%. Data are expressed as mean ± SEM. **p < 0.01 or ***p < 0.001 versus control group, # p < 0.05, ## p < 0.01 or ### p < 0.001 versus H2O2 treatment group
SN Exhibited Little Free Radical Scavenging Property In Vitro
Then, we investigated the mechanism underlying the protection of SN against oxidative stress. One possible explanation is that the intracellular SN exerts direct free radical scavenging property. To confirm this hypothesis, we studied the antioxidant effect of SN in vitro. As shown in Fig. 3, the hydroxyl radical (OH•) was produced by the Fenton reaction and the levels of OH• were evaluated by electron spin resonance (ESR) measurement. As compared to control group, cotreatment with different concentrations of SN (0.1, 0.5, 1, or 5 μM) could not significantly decrease the signal intensity of PBN-OH adducts (n = 5), which suggested that SN exhibited little direct effects on scavenging ROS.

SN exhibited little direct free radical scavenging property in vitro. ESR spectra of the spin adduct of OH• radical were observed during the reaction of 0.5 mM H2O2 containing 30 mM PBN and 0.2 mM FeSO4 with or without indicated concentrations of SN (0.1, 0.5, 1, and 5 μM). The levels of ESR peak intensity were expressed as a relative change in comparison with control, which was set to 100%. Data are expressed as mean ± SEM
SN Pre-conditioning Dose-Dependently Increased the Expression of Nrf2-related Antioxidant Genes, Which Mediated the Protective Effects of SN
Nrf2 antioxidant system is considered as good candidate molecules for modulating intracellular redox state. We hypothesized that SN pre-conditioning may increase the expression of Nrf2-related antioxidant genes in PC12 cells. Firstly, we analyzed the relative expression of Nrf2-related antioxidant genes including Nrf2, HO-1, and NQO-1 by quantitative real-time PCR. As shown in Fig. 4a–c, compared to the mRNA expression in the control group, the Nrf2 (n = 5, *p < 0.05 or ***p < 0.001 vs. control, Fig. 4a), HO-1 (n = 5, ***p < 0.001 vs. control, Fig. 4b), and NQO-1 mRNA (n = 5, **p < 0.01 or ***p < 0.001 vs. control, Fig. 4c) expression was significantly increased in a dose-dependent manner when PC12 cells were pre-incubated with SN at a concentration of 1, 5, 10, and 50 μM for 6 h. Next, we investigated the effect of SN on the protein expression of Nrf2 antioxidant system. It was found that pretreatment with SN (5 μM, 24 h) obviously elevated the protein expression of Nrf2, HO-1, and NQO-1 to 215.12 ± 33.43% (n = 5, **p < 0. 01 vs. control, Fig. 4d), 289.23 ± 42.70% (n = 5, ***p < 0.001 vs. control, Fig. 4d), and 172.23 ± 15.32% (n = 5, *p < 0.05, Fig. 4d), respectively, when compared with the control group. Moreover, in order to confirm the role of Nrf2 in the neuroprotective effects of SN, we compared its actions on the rate of cell viability following H2O2 exposure in control and Nrf2-silenced PC12 cells. Expectedly, the protective effects of SN were largely abolished when Nrf2 was silenced (n = 5, $ p < 0.05 vs. H2O2 + scrambled-siRNA, Fig. 4e).

SN dose-dependently increased the expression of Nrf2-related antioxidant genes, which mediated the protective effects of SN. a–c PC12 cells were treated with SN (1, 5, 10, or 50 μM) for 6 h. The mRNA expression of Nrf2 (a), HO-1 (b), and NQO-1 (c) was evaluated by quantitative real-time PCR. Res was used as a positive control. d PC12 cells were treated with SN (5 μM) for 24 h. The protein expression of Nrf2, HO-1, and NQO-1 was evaluated by western blotting. e PC12 cells were transfected with scrambled-siRNA or Nrf2-siRNA and incubated with H2O2 (200 μM) for 12 h. The effects of Nrf2 silencing on the protective effects of SN were evaluated by MTT assay. Results were expressed as a relative change in comparison with controls, which was set to 100%. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01 or ***p < 0.001 versus control group, ### p < 0.001 versus H2O2 treatment group, $ p < 0.05 versus H2O2 + scrambled-siRNA group
SN Increased Intracellular ROS Levels at its High Concentration
We therefore asked the underlying mechanism that how SN increased the expression of Nrf2-related antioxidant genes. Although overproduction of ROS causes cell injury, ROS at low concentration may provide beneficial effects via triggering cellular endogenous antioxidant system (Fourquet et al. 2010). It is well accepted that morphine concentration and time dependently generate cellular ROS, which is proverbially involved in the effect of morphine (Ma et al. 2015). SN belongs to the family of dextrorotatory morphinan analogues; therefore, we speculated that SN activated the Nrf2 signaling system through induction of intracellular “appropriate” production of ROS. As shown in Fig. 5, the levels of MDA, the content of GSH and the activity of SOD were assessed when PC12 cells were treated with higher concentrations of SN (1, 5, 10 and 50 μM). SN at a concentration of 1, 5 or 10 μM did not efficiently alter the levels of MDA (Fig. 5a), the content of GSH (Fig. 5b) and SOD activity (Fig. 5c). However, the content of MDA was efficiently elevated (n = 5, *p < 0.05 vs. control, Fig. 5a) and the activity of SOD was significantly reduced (n = 5, *p < 0.05 vs. control, Fig. 5c) when PC12 cells were treated with 50 μM SN for 1 h. These data suggested that SN increased intracellular ROS levels at its high concentration, which indicated that the SN-induced activation of Nrf2-antioxidant system may be ROS-dependent.

SN increased intracellular ROS levels at its high concentration. PC12 cells were incubated with different concentrations of SN (1, 5, 10, and 50 μM) for 1 h. The levels of MDA (a), the content of GSH (b), and the activity of SOD (c) were evaluated. Results were expressed as a relative change in comparison with controls, which was set to 100%. Data are expressed as mean ± SEM. *p < 0.05 versus control group
The antioxidant trolox significantly prevented the SN-induced up-regulation of Nrf2-related antioxidant genes
To further confirm that SN increased the expression of Nrf2-related antioxidant genes via a ROS-dependent mechanism, we studied the effect of the antioxidant trolox on the SN-induced activation of Nrf2 signaling system. As shown in Fig. 6a–c, pretreatment with SN (1 and 5 μM, 6 h) obviously increased the mRNA expression of Nrf2, HO-1 and NQO-1 (n = 5, *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control) in a dose-dependent manner. Similar results were obtained in primary neurons (n = 5, *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control Fig. 6d–f). However, when the antioxidant trolox was co-treated with SN in PC12 cells, the SN-induced dose-dependent augment in the expression of Nrf2 (n = 5, # p < 0.05 vs. 1 μM SN and $ p < 0.05 vs. 5 μM SN, Fig. 6a), HO-1 (n = 5, ## p < 0.01 vs. 1 μM SN and $$ p < 0.01 vs. 5 μM SN, Fig. 6b), and NQO-1 (n = 5, # p < 0.05 vs. 1 μM SN and $ p < 0.05 vs. 5 μM SN, Fig. 6c) was significantly attenuated. These data indicated that SN activated Nrf2 antioxidant system via triggering ROS generation, followed by the increase of cell resistance to oxidative stress.

The antioxidant trolox significantly prevented the SN-induced up-regulation of Nrf2-related antioxidant genes. a–c PC12 cells were co-incubated with SN (1 or 5 μM) and Trolox (0 or 0.5 mM) for 6 h. The mRNA expression of Nrf2 (a), HO-1 (b), and NQO-1 (c) was evaluated by quantitative real-time PCR. d–f Primary cultured cortical neurons were treated with SN (1 and 5 μM) for 6 h. The mRNA expression of Nrf2 (d), HO-1 (e), and NQO-1 (f) was evaluated by quantitative real-time PCR. Results were expressed as a relative change in comparison with controls, which was set to 100%. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01 or ***p < 0.001 versus control group, # p < 0.05 or ## p < 0.01 versus 1 μM SN treatment group, $ p < 0.05 or $$ p < 0.01 versus 5 μM SN group
SN Pre-conditioning Inhibited the Activation of NOX
Previous studies have showed that SN exhibits anti-inflammatory effects through inhibiting the activation of NADPH oxidase (NOX) in the microglia; therefore, we asked if there exist other mechanisms involved in the protective role of SN against H2O2-induced cytotoxicity and oxidative stress. As shown in Fig. 7, the NOX2 expression (n = 5, *p < 0.05 vs. control, Fig. 7a) and the activity of NOX (n = 5, **p < 0.01 vs. control, Fig. 7b) were markedly increased in the H2O2-treated PC12 cells compared to control cells. Although pretreatment with SN at a concentration of 5 μM had no significant effect on the H2O2-induced protein expression of NOX2 (n = 5, Fig. 7a), the increase of NOX activity induced by H2O2 was obviously attenuated (n = 5, # p < 0.05 vs. control, Fig. 7b).

SN pre-conditioning inhibited the activation of NOX. PC12 cells were pre-incubated 24 h with or without SN (5 μM) prior to H2O2 (200 μM) exposure for another 24 h. a The protein expression of NOX2 was evaluated by western blotting. b The NOX activity was detected by lucigenin-enhanced chemiluminescence with colorimetric assay kit. Results were expressed as a relative change in comparison with controls, which was set to 100%. Data are expressed as mean ± SEM. *p < 0.05 or **p < 0.01 versus control group, # p < 0.05 versus H2O2 treatment group
Discussion
In this work, we report for the first time that pretreatment with SN, a natural alkaloid extracted from Chinese medicinal plant S. acutum extensively used in treatment of rheumatoid arthritis and various inflammatory disorders, up-regulated the endogenous antioxidant system including Nrf2, HO-1, and NQO-1, in PC12 cells via a ROS-dependent mechanism. This mechanism was mainly responsible for the protective effects of SN pretreatment against cellular oxidative stress, for SN exhibited little direct scavenging effect in a Fenton reaction system. Evidence collected in this study provided a new mechanism underlying the neuroprotective effects of SN.
Previous studies have showed that SN displays adverse spectrum of biological activities, including anti-inflammatory, neuroprotective, and immune suppressive potential (Chen et al. 2011; Feng et al. 2006; Mark et al. 2003; Qian et al. 2007). Considering its ability to come across blood–brain barrier (Liu et al. 2005; Long et al. 2010), and increasing evidence for its beneficial effects on central nerve system, SN emerged as a new pharmacological agent to rescue neurobiological diseases, including stroke and PD (Qian et al. 2007; Wu et al. 2011). The generation of ROS is an essential metabolic process for maintaining homeostasis. A series of antioxidant mechanisms maintain intracellular redox homeostasis. When they deficit, overproduction of ROS causes DNA mutations and protein dysfunction, which eventually leads to neuronal injury and neurobiological diseases (Espinosa-Diez et al. 2015; Schieber and Chandel 2014; Zhang et al. 2011). We used PC12 neuronal cells, which share many properties with primary sympathetic neurons and chromaffin in cell cultures, to evaluate the effect of SN on ROS. We found that SN exhibited little effects on scavenging the exogenous ROS, but pre-incubation with SN significantly increased the resistance to oxidative stress in PC12 cells. The indirect antioxidant effect of SN may underlay the beneficial effects of SN in the central nerve system.
A serious of studies has found that SN exhibited antioxidant effects in different cells, including neuronal cells (Li et al. 2008; Qin et al. 2016b; Wang et al. 2016; Yang et al. 2014). However, to our knowledge, there are fewer reports about the mechanism underlying the antioxidant effect. Previous studies have revealed that SN inhibited the activation of NADPH oxidase (NOX) in the microglia, which may confer to the anti-inflammation of SN (Qian et al. 2007). In our study, we used an oxidative stress model by addition of exogenous hydrogen peroxide, which is not entirely dependent on NOX. Interestingly, it was observed that pre-incubation with SN exhibited a good protection against cellular oxidative stress. Meanwhile, we also found that the beneficial effects of SN at low concentration (1 μM) were abolished when SN was co-incubated with H2O2. However, 1 μM SN pretreatment exhibited significant protective effects against oxidative stress, indicating that SN may trigger the resistance to oxidative stress in PC12 cells. We further observed the effects of SN pretreatment on the NOX2 expression and the activity of NOX in PC12 cells exposure to oxidative stress. Little effect of SN pretreatment on the expression of NOX2 in PC12 cells was observed. The activity of NOX was reduced by SN pretreatment at high concentration (5 μM). But at low concentration (1 μM), SN pretreatment exhibited significant protective effects against oxidative stress, without inhibition on NOX activity in PC12 cells. Thus, the effects of SN pretreatment at low concentration against oxidative stress might not be attributed to inhibition of NOX activity. There may exist other mechanisms underlying its antioxidant effects. This mechanism was not due to its direct radical scavenging property, for SN was removed from the medium before the exogenous hydrogen peroxide was added. Furthermore, we observed that SN exhibited little direct effects on scavenging the exogenous ROS. Nrf2 is known to be an important transcription factor that is involved in the cellular antioxidant response, as it binds to antioxidant response elements (ARE) in the genes encoding antioxidant enzymes, such as HO-1 and NQO-1 (Buendia et al. 2016; Hybertson and Gao 2014). Moreover, it has also been reported that SN activated Nrf2 in the kidney (Qin et al. 2016a). Thus, we observed the effects of SN on the Nrf2 system in PC12 cells. We found that after incubation with SN, the mRNA level and the protein level of Nrf2 system in PC12 cells, including Nrf2, HO-1, and NQO-1, were up-regulated in a concentration-dependent manner. Moreover, the protective effects of SN pretreatment on PC12 cells were largely attenuated when Nrf2 was silenced. Our findings mainly explained the antioxidant effects of SN pretreatment on cells.
An interesting question is that how could SN activate Nrf2 system? Based on its molecular structure, SN belongs to the family of dextrorotatory morphinan analogues (Jin et al. 2008). It is well known that morphine generates cellular ROS in a concentration- and time-dependent manner and ROS is proverbially involved in the effect of morphine (Ma et al. 2015). Although overproduction of ROS causes cell injury, ROS at low concentration may provide beneficial effects via triggering cellular endogenous antioxidant system (Fourquet et al. 2010). Interestingly, we found that SN increased intracellular ROS levels at its high concentration, indicating that SN may up-regulate endogenous antioxidant system via a ROS-dependent pathway, which is further confirmed by the result that incubation with antioxidant trolox prevented the up-regulation of Nrf2 system induced by SN. This point was in according with previous reports that morphine protected SH-SY5Y human neuroblastoma cells against 6-OHDA-induced cell injury via increasing cellular antioxidant (Elyasi et al. 2014). It should be noted that although it seemed that the lower concentrations of SN did not increase ROS level but activated Nrf2 in PC12 cells, the effects of SN pretreatment on PC12 cells were abolished by trolox, a ROS scavenger. We hypothesized that the increased ROS level of SN pretreatment with low concentration was not observed because of the detection limitation. Altogether, these evidences strongly supported that SN increased the resistance to oxidative stress in PC12 cells via triggering beneficial levels of ROS and activating the cellular antioxidant response. However, there may exist other potential mechanisms. For instance, SN shows a Michel acceptor on the structure that might react with the cysteine residues present at keap1 that might be involved in the induction capability. This possibility required further investigation.
In summary, this study showed that pre-incubation with SN at low concentration induced cytoprotection against oxidative stress-induced neuronal cell death via triggering an “appropriate” ROS signal, followed by activation of cellular defense system. As a clinic-used drug in the therapy of inflammatory disorders, SN may be used not only in the treatment but also in the prevention of neurological diseases in the future.
References
Bao GH, Qin GW, Wang R, Tang XC (2005) Morphinane alkaloids with cell protective effects from Sinomenium acutum. J Nat Prod 68:1128–1130. doi:10.1021/np050112+
Buendia I, Michalska P, Navarro E, Gameiro I, Egea J, Leon R (2016) Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther 157:84–104. doi:10.1016/j.pharmthera.2015.11.003
Carvalho AN, Firuzi O, Gama MJ, van Horssen J, Saso L (2016) Oxidative stress and antioxidants in neurological diseases: is there still hope? Current drug targets. doi:10.2174/1389450117666160401120514
Chen DP et al (2011) Anti-inflammatory activities of Chinese herbal medicine sinomenine and Liang Miao San on tumor necrosis factor-alpha-activated human fibroblast-like synoviocytes in rheumatoid arthritis. J Ethnopharmacol 137:457–468. doi:10.1016/j.jep.2011.05.048
Elyasi L, Eftekhar-Vaghefi SH, Esmaeili-Mahani S (2014) Morphine protects SH-SY5Y human neuroblastoma cells against 6-hydroxydopamine-induced cell damage: involvement of anti-oxidant, calcium blocking, and anti-apoptotic properties. Rejuvenation Res 17:255–263. doi:10.1089/rej.2013.1473
Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sanchez-Perez P, Cadenas S, Lamas S (2015) Antioxidant responses and cellular adjustments to oxidative stress redox biology 6:183–197. doi:10.1016/j.redox.2015.07.008
Fan H et al (2015) Methionine sulfoxide reductase A negatively controls microglia-mediated neuroinflammation via inhibiting ROS/MAPKs/NF-kappaB signaling pathways through a catalytic antioxidant function. Antioxid Redox Signal 22:832–847. doi:10.1089/ars.2014.6022
Feng H, Yamaki K, Takano H, Inoue K, Yanagisawa R, Yoshino S (2006) Suppression of Th1 and Th2 immune responses in mice by Sinomenine, an alkaloid extracted from the chinese medicinal plant Sinomenium acutum. Planta Med 72:1383–1388. doi:10.1055/s-2006-951721
Fourquet S, Guerois R, Biard D, Toledano MB (2010) Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem 285:8463–8471. doi:10.1074/jbc.M109.051714
Hybertson BM, Gao B (2014) Role of the Nrf2 signaling system in health and disease. Clin Genet 86:447–452. doi:10.1111/cge.12474
Jin HZ, Wang XL, Wang HB, Wang YB, Lin LP, Ding J, Qin GW (2008) Morphinane alkaloid dimers from Sinomenium acutum. J Nat Prod 71:127–129. doi:10.1021/np0704654
Kim GH, Kim JE, Rhie SJ, Yoon S (2015) The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol 24:325–340. doi:10.5607/en.2015.24.4.325
Li L, Gao XL, Ding BX (2008) Inhibitory effect of sinomenine on H2O2-induced apoptosis in neonatal rat cardiomyocytes. Zhongguo Zhong yao za zhi = Zhongguo zhongyao zazhi = China journal of Chinese materia medica 33(939–941):961
Li J, Zhao L, He X, Zeng YJ, Dai SS (2013) Sinomenine protects against lipopolysaccharide-induced acute lung injury in mice via adenosine A(2A) receptor signaling. PLoS ONE 8:e59257. doi:10.1371/journal.pone.0059257
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795. doi:10.1038/nature05292
Liu ZQ, Chan K, Zhou H, Jiang ZH, Wong YF, Xu HX, Liu L (2005) The pharmacokinetics and tissue distribution of sinomenine in rats and its protein binding ability in vitro. Life Sci 77:3197–3209. doi:10.1016/j.lfs.2005.05.054
Long LH et al (2010) HPLC and LC-MS analysis of sinomenine and its application in pharmacokinetic studies in rats. Acta Pharmacol Sin 31:1508–1514. doi:10.1038/aps.2010.122
Ma J, Yuan X, Qu H, Zhang J, Wang D, Sun X, Zheng Q (2015) The role of reactive oxygen species in morphine addiction of SH-SY5Y cells. Life Sci 124:128–135. doi:10.1016/j.lfs.2015.01.003
Mark W et al (2003) Sinomenine blocks tissue remodeling in a rat model of chronic cardiac allograft rejection. Transplantation 75:940–945. doi:10.1097/01.TP.0000056610.22062.03
Popa-Wagner A, Mitran S, Sivanesan S, Chang E, Buga AM (2013) ROS and brain diseases: the good, the bad, and the ugly. Oxid Med Cell Longev 2013:963520. doi:10.1155/2013/963520
Qian L, Xu Z, Zhang W, Wilson B, Hong JS, Flood PM (2007) Sinomenine, a natural dextrorotatory morphinan analog, is anti-inflammatory and neuroprotective through inhibition of microglial NADPH oxidase. J neuroinflammation 4:23. doi:10.1186/1742-2094-4-23
Qin T, Du R, Huang F, Yin S, Yang J, Qin S, Cao W (2016a) Sinomenine activation of Nrf2 signaling prevents hyperactive inflammation and kidney injury in a mouse model of obstructive nephropathy. Free Radic Biol Med 92:90–99. doi:10.1016/j.freeradbiomed.2016.01.011
Qin T, Yin S, Yang J, Zhang Q, Liu Y, Huang F, Cao W (2016b) Sinomenine attenuates renal fibrosis through Nrf2-mediated inhibition of oxidative stress and TGFbeta signaling. Toxicol Appl Pharmacol 304:1–8. doi:10.1016/j.taap.2016.05.009
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24:R453–R462. doi:10.1016/j.cub.2014.03.034
Wang W et al (2016) Sinomenine attenuates Angiotensin II-induced autophagy via inhibition of P47-Phox translocation to the membrane and influences reactive oxygen species generation in podocytes. Kidney Blood Press Res 41:158–167. doi:10.1159/000443417
Wu WN et al (2011) Sinomenine protects against ischaemic brain injury: involvement of co-inhibition of acid-sensing ion channel 1a and L-type calcium channels. Br J Pharmacol 164:1445–1459. doi:10.1111/j.1476-5381.2011.01487.x
Xu M, Liu L, Qi C, Deng B, Cai X (2008) Sinomenine versus NSAIDs for the treatment of rheumatoid arthritis: a systematic review and meta-analysis. Planta Med 74:1423–1429. doi:10.1055/s-2008-1081346
Yang Z, Liu Y, Yuan F, Li Z, Huang S, Shen H, Yuan B (2014) Sinomenine inhibits microglia activation and attenuates brain injury in intracerebral hemorrhage. Mol Immunol 60:109–114. doi:10.1016/j.molimm.2014.03.005
Zhang Y, Du Y, Le W, Wang K, Kieffer N, Zhang J (2011) Redox control of the survival of healthy and diseased cells. Antioxid Redox Signal 15:2867–2908. doi:10.1089/ars.2010.3685
Acknowledgement
This work was supported by the National Nature Science Foundation of China (Grant Nos. U1504808 and 81501075).
Author Contributions
Hua Fan, Qing Shu, and Juan Li conceived and designed the experiments; Xinlei Guan, Jiegang Zhao, Junqiang Yan, Xiangming Li, Jiangbo Liu, Zhaohui Jia, and Jian Shi performed the experiments; Xinlei Guan and Qing Shu analyzed the data; Hua Fan and Juan Li wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Hua Fan, Qing Shu and Xinlei Guan have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Fan, H., Shu, Q., Guan, X. et al. Sinomenine Protects PC12 Neuronal Cells against H2O2-induced Cytotoxicity and Oxidative Stress via a ROS-dependent Up-regulation of Endogenous Antioxidant System. Cell Mol Neurobiol 37, 1387–1398 (2017). https://doi.org/10.1007/s10571-017-0469-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-017-0469-1