
ABSTRACT
Growth arrest-specific 1 (Gas1) protein acts as an inhibitor of cell growth and a mediator of cell death in nervous system during development and is also re-expressed in adult neurons during excitotoxic insult. Due to its structural similarity to the glial cell-derived neurotrophic factor family receptors α (GFRα), Gas1 is likely to interfere with the neuroprotective effect of GDNF. In the present study, we investigated the expression profile of Gas1 during glutamate insults in human SH-SY5Y neuroblastoma cells as well as the influence of Gas1 inhibition on the protective effect of GDNF against glutamate-induced cell injury. Our data showed that Gas1 expression was significantly increased with the treatment of glutamate in SH-SY5Y cells. The silencing of Gas1 by small interfering RNA promoted the protective effect of GDNF against glutamate-induced cytotoxicity as well as cell apoptosis, which effect was likely mediated through activating Akt/PI3 K-dependent cell survival signaling pathway and inhibiting mitochondrial-dependent cell apoptosis signaling pathway via Bad dephosphorylation blockade. In summary, this study showed the synergistic effect of Gas1 inhibition and GDNF against glutamate-induced cell injury in human SH-SY5Y neuroblastoma cells, which information might significantly contribute to better understanding the function of Gas1 in neuronal cells and form the basis of the therapeutic development of GDNF in treating human neurodegenerative diseases in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With an increasing aging population, neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntingson’s disease (HD) are becoming more prevalent world widely (Pizza et al. 2011). Most neurodegenerative diseases are resulted from the loss of structure or function of neurons in the brain and spinal cord (Gibson et al. 2010; Saxena and Caroni 2011). In neurodegenerative disorders, the primary causes of neuronal cell death include excitotoxicity, oxidative stress, apoptosis, and protein deposition (e.g., α-synuclein, β-amyloid) (Beal 1992; Ueda et al. 1994, Yuan et al. 2003). Glutamate, a major excitatory amino acid neurotransmitter naturally occurring in the central nervous system (CNS), mediates several essential physiological processes (Greenamyre and Porter 1994). However, the excess release of glutamate in the CNS significantly contributes to these neurological disorders (Coyle and Puttfarcken 1993). A few studies have elucidated that glutamate induces excitotoxicity in primary cultured neurons through both oxidative damage and over-stimulation of N methyl-d-aspartate (NMDA) receptors, the latter of which would lead to calcium homeostasis destruction (Ankarcrona et al. 1995). Moreover, a second wave of gene expression alternation followed by glutamate injury was reported to be involved in excitotoxic neuronal death (Wang and Qin 2010). Growth arrest-specific gene 1 (Gas1), a glycosylphosphatidylinositol (GPI)-anchored protein, is widely expressed in the nervous system and plays an important role in growth inhibition and cell death induction during development (Del Sal et al. 1992; Zarco et al. 2012, 2013). Aberrant Gas1 expression was found in adult neurons during excitotoxicity, which is potentially associated with pro-apoptotic effects (Mellstrom et al. 2002).
Several regimens targeting at the neurodegenerative diseases have provided new options for the prevention and treatment of such diseases (Chalak and Rouse 2011). Neuronal growth factors have been widely taken into consideration, given their roles in promoting neuronal survival in the developing nervous system (Edgar and Barde 1983). Glial cell line-derived neurotrophic factor (GDNF), a classic member of neurotrophic factors (NTFs) identified in conditioned media from a glial cell line, is prioritized as one of neuroprotective factors due to its ability in promoting the survival of mesencephalic dopaminergic neurons (Lin et al. 1993; Yan et al. 1995). In the adult brain, target-derived GDNF is required for the maintenance of dopaminergic neurons and protection against injury (Choi-Lundberg et al. 1997). Arising in vitro and in vivo evidence has indicated that GDNF has the protective effect against the excitotoxic damage in neurons (Bonde et al. 2000; Gratacòs et al. 2001); however, clinical trails with application of GDNF to neurodegenerative diseases such as PD show inconsistent results (Lang et al. 2006). Several explanations for this difference have been proposed, but until now, the detailed molecular and cellular mechanism underpinning such effect still remains unclear. It has been postulated that GDNF binds to its cognate co-receptor glial cell-derived neurotrophic factor family receptors α (GFRα), specifically activates Ret kinase and thus, prevents neuronal injury (Treanor et al. 1996). Interestingly, Gas1 exhibits significant structural homology with GFRα and acts as an negative modulator of GDNF signaling (Schueler-Furman et al. 2006; López-Ramírez et al. 2008).
In this study, we investigated the expression profile of Gas1 during glutamate insults and evaluated the influence of Gas1 inhibition on the protective effect of GDNF against glutamate-induced cell injury in human SH-SY5Y neuroblastoma cells, which cell line was reported to express GFRα1 and Ret. Furthermore, we also explored the molecular events during such cellular process.
Materials and Methods
Materials
Recombinant human GDNF was purchased from PeproTech (NJ, USA). Growth medium components were purchased from Gibco (NY, USA). L-glutamate, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (MO, USA). Annexin V-FITC and PI double staining kit was obtained from BD Biosciences (CA, USA). Antibodies used in this study were purchased from Santa Cruz Biotechnology (CA, USA) and Cell Signaling Technology (MA, USA). Caspase-3,9 fluorometric assay kits were obtained from BioVision (SF, USA). All chemicals and reagents were of analytical grade.
Cell Culture
Human SH-SY5Y neuroblastoma cells were obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). Cells were maintained in Minimum Essential Medium (MEM)/F12 medium, supplemented with 1 % non-essential amino acids, 10 % fetal bovine serum (FBS), 100 U/mL penicillin, and 100 U/mL streptomycin at 37 °C in 5 % CO2. The cells were passaged once every 3 days.
Gas1 Silencing
Gene silencing of Gas1 using small interfering RNA (siRNA) was achieved as described previously (López-Ramírez et al. 2008). The Gas1-specific siRNA (siRNA-Gas1) was purchased from Santa Cruz Biotechnology (Cat. No: sc-37435, CA, USA). A non-targeting scrambled siRNA served as non-specific control (siRNA-C, Cat. No: sc-37007). Cells were transfected with siRNA (50 nM) in serum-free medium for 5 h using Metafectene reagent (Biontex, München, Germany) according to the manufacturer’s protocol. The depletion of target gene was determined by real-time polymerase chain reaction (RT-PCR) and Western blot analysis.
Real-Time PCR Analysis
After treatment, total RNA was extracted using Trizol reagent from cells according to the manufacturer’s instructions. First-strand cDNA was synthesized using PrimeScript™ RT reagents Kit (Takara, Shiga, Japan). RT-PCR was performed with the cDNA using SYBR green PCR Master Mix (Takara, Shiga, Japan). Each amplification reaction underwent denaturation at 95 °C for 30 s, amplification for 40 cycles at 95 °C for 5 s, annealing and extension at 60 °C for 20 s using ABI7500 sequence detection system (Life Technologies, ON, USA). In all experiments, β-actin was used as the housekeeping gene for gene expression normalization.
Western Blot Analysis
After treatment, cells were collected and lysed in lysis buffer (10 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1 % sodium dodecyl sulfate, 1 % Triton X-100, that contained the protease inhibitors phenylmethylsulfonyl fluoride, 200 mg/mL, and leupeptin, 3 mg/mL, pH 7.4). Bradford assay was used to measure protein concentration (Kruger 1994). Then protein samples were electrophoresed on 12 % SDS-polyacrylamide gel (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. Each membrane was incubated for 1 h with 5 % bovine serum albumin (BSA) in PBS-Tween at room temperature and then probed with primary antibody at 4 °C overnight. On the next day, the membrane was washed with PBS-Tween, incubated with secondary antibody for one hour at room temperature. The membrane was washed with PBS-Tween and then visualized using the enhanced chemiluminescence (ECL) assay kit (Beyotime, Nantong, China). In all experiments, membranes were re-probed for β-actin and the density of each band was normalized to the expression of β-actin.
MTT Assay
Cell viability was measured using MTT assay as previously reported (van Meerloo et al. 2011). Briefly, after treatment, 10 μL MTT (1 mg/mL) was added to each well and incubated for 4 h at 37 °C. After incubation, the culture medium was removed and DMSO (100 μL) was added to each well to dissolve the formazan crystals. The absorbance was read with a microplate reader (Molecular Devices, CA, USA) at 570 nm.
Flow Cytometric Analysis
The presence of apoptotic cells was determined by flow cytometric analysis using Annexin V-FITC and PI apoptosis kit. Briefly, after treatment, cells were re-suspended in 300-μL binding buffer containing 10 μL of Annexin V-FITC stock and 10 μL of PI, and then incubated at room temperature for 15 min in the dark. The samples were analyzed by a flow cytometer (Becton–Dickinson, CA, USA). The percentages of apoptotic cells were estimated for each sample.
Cytochrome c Release Measurement
For measurement of cytochrome c release, the cytosol and mitochondrial fractions were prepared as described previously (Zhu et al. 2014). After treatment, mitochondrial and cytosolic fractions were extracted from the cells using Apo Alert Cell Fractionation Kit (Clontech, CA, USA) according to the manufacturer’s instructions. The expression of cytochrome c was determined using a monoclonal antibody through Western blot analysis as described before.
Caspase Activity Assay
After treatment, cells were collected and lysed in caspase assay buffer containing 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (pH 7.5), 100 mM NaCl, 2 mM EDTA (ethylene diamine tetraacetic acid), 0.1 % CHAPS, 10 % sucrose, and 5 mM DTT (dithiothreitol). Aliquots of crude cell lysate were incubated with caspase-3 substrate DEVD-AFC or caspase-9 substrate LEHD-AFC at 37 °C for 30 min. The caspase activity was determined by measuring the relative fluorescence intensity at 505 nM following excitation at 400 nm using a spectrofluorometer (Molecular Devices, CA, USA).
Statistical Analysis
Biostatistical analyses were conducted with the Prism 5.0 and SPSS 16.0 software packages. All experiments were repeated three times. Results of multiple experiments were expressed as mean ± SD. Comparisons between experimental and control groups were performed by Student’s t test and one-way ANOVA and differences were considered to be statistically significant when p value was less than 0.05.
Results
Expression of Gas1 in SH-SY5Y Cells with Glutamate Treatment
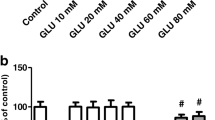
As reported previously, SH-SY5Y cells expressed high levels of GFRα1 and Ret, but very low or undetectable level of Gas1 when cultured in serum. However, after serum withdrawal, the expression of Gas1 was detectable in cells (Cabrera et al. 2006). In the current study, cells were treated with glutamate (10 mM) for different time periods and then the levels of related receptors were analyzed. Our results showed that exposure to glutamate had no effect on the expression of GFRα1 and Ret (data not shown), but resulted in a rapid accumulation of Gas1 protein in cells that was detectable as early as 6 h and then reached a plateau between 12 and 16 h (Fig. 1). This finding suggested Gas1 was involved in glutamate-induced excitotoxic death in human SH-SY5Y neuroblastoma cells.

Gas1 expression in SH-SY5Y cells with the treatment of glutamate (10 mM) for different time periods. a A representative experiment of the expression of Gas1 protein in SH-SY5Y cells treated with glutamate (10 mM) at different times. b The densitometric analysis of the expression of Gas1 protein in SH-SY5Y cells treated with glutamate (10 mM) at different times. The results were shown as mean ± SD of three experiments with triplicate sets in each experiment. **p < 0.01 versus treatment with glutamate at 6 h
Gas1 Silencing Increases the Protective Effect of GDNF Against Glutamate-Induced Cytotoxicity
GDNF was reported to selectively attenuate excitotoxin-induced neuronal death; however, this effect was not pronounced (Bonde et al. 2000). Since Gas1 acted as a negative modulator of GDNF signaling, we then investigated whether Gas1 blockade could promote the protective effect of GDNF against glutamate-induced cytotoxicity. We first confirmed that GDNF treatment could not induce the expression of Gas1 in SH-SY5Y cells (data not shown). Taking advantage of the small interfering RNA technology, Gas1 silencing was achieved at both mRNA and protein levels under glutamate treatment (Fig. 2). Cell viability of SH-SY5Y cells treated with or without indicated drugs was assessed with MTT assay. As shown in Fig. 3, GDNF (50 ng/mL) exhibited a moderately protective effect on the growth of SH-SY5Y cells against glutamate-induced neurotoxicity. Such effect was pronouncedly increased in Gas1 knockdown cells compared to that in control. Our results indicated that Gas1 silencing could markedly enhance GDNF’s protective effect against glutamate-induced cytotoxicity in human SH-SY5Y neuroblastoma cells.

Gas1 silencing in SH-SY5Y cells with glutamate treatment. SH-SY5Y cells with or without Gas1 knockdown were treated with glutamate (10 mM) for 16 h and the relative mRNA and protein expressions of Gas1 were assessed by RT-PCR (a) and Western blot analysis (b). A non-targeting scrambled siRNA was used as the negative control in these experiments. The results were shown as mean ± SD of three experiments with triplicate sets in each experiment. **p < 0.01 versus control, ## p < 0.01 versus treatment with glutamate alone in SH-SY5Y cells

Gas1 silencing promoted the protective effect of GDNF against glutamate-induced cytotoxicity. SH-SY5Y cells with or without Gas1 knockdown were treated with glutamate (10 mM) in the absence or presence of GDNF (50 ng/mL) for 24 h and cell viability was then assessed with MTT assay. The results were shown as mean ± SD of three experiments with triplicate sets in each experiment. *p < 0.05 versus control, # p < 0.05 versus treatment with glutamate alone in SH-SY5Y cells, ^ p < 0.05 versus treatment with glutamate and GDNF in SH-SY5Y cells
Gas1 Silencing Increases the Protective Effect of GDNF Against Glutamate-Induced Apoptosis
The previous study showed that GDNF attenuated excitotoxin-induced neuronal death, primarily through reducing cell apoptosis (Ghribi et al. 2001). Gas1 is known to be an inhibitor of cell growth and a mediator of cell death in many different types of cells (Del Sal et al. 1992; Evdokiou and Cowled 1998). Apoptosis of SH-SY5Y cells treated with or without indicated drugs was evaluated by dual-staining with Annexin V-FITC/PI. As shown in Fig. 4, treatment with glutamate (10 mM) remarkably increased the percentage of early apoptotic cells, while co-treatment with GDNF (50 ng/mL) resulted in a decreased cell apoptosis from 52.35 % ± 3.47 % to 43.26 ± 2.28 %. Furthermore, Gas1 silencing increased the protective effect of GDNF with a decreased apoptosis from 52.35 % ± 3.47 % to 22.18 % ± 4.82 %. Our data suggested that the synergistic protective effect of Gas1 silencing and GDNF was also mediated through inhibiting glutamate-induced cell apoptosis in human SH-SY5Y neuroblastoma cells.

Gas1 silencing increases the protective effect of GDNF against glutamate-induced apoptosis. SH-SY5Y cells with or without Gas1 knockdown were treated with glutamate (10 mM) in the absence or presence of GDNF (50 ng/mL) for 24 h. a Flow cytometry analysis of cell apoptosis using Annexin V-FITC/PI dual-staining. b The densitometric analysis of the percentage distribution of apoptotic cells. The results were shown as mean ± SD of three experiments with triplicate sets in each experiment. **p < 0.01 versus control, ## p < 0.01 versus treatment with glutamate alone in SH-SY5Y cells, ^^ p < 0.01 versus treatment with glutamate and GDNF in SH-SY5Y cells
Gas1 Silencing Promotes the Protective Effect of GDNF via the Activation of PI3 K/Akt-Dependent Cell Survival Signaling Pathway
Previous report showed that Gas1 could inhibit cell survival by blocking the GDNF-mediated signaling pathway (López-Ramírez et al. 2008). In this study, we found that GDNF exerted its protective effect against glutamate injury by inducing Ret phosphorylation in SH-SY5Y cells; furthermore, this effect was more pronounced in SH-SY5Y cells with Gas1 knockdown (Fig. 5). By following the downstream intracellular signaling cascade, we observed that the phosphorylations of Akt and Bad, two downstream targeted genes of Ret, were significantly decreased with the treatment of glutamate; however, those were increased by the co-treatment of GDNF. The up-regulated expression of p-Akt and p-Bad was more pronounced in Gas1 knockdown cells. Our results suggested Gas1 silencing promoted GDNF’s protective effect via activating PI3 K/Akt-dependent cell survival signaling pathway.

Gas1 silencing promotes the protective effect of GDNF against glutamate-induced cytotoxicity through modulating the PI3 K/Akt-dependent survival signaling pathways. SH-SY5Y cells with or without Gas1 knockdown were treated with glutamate (10 mM) in the absence or presence of GDNF (50 ng/mL) for 24 h. a The expression of related proteins was assessed by Western blot analysis. b The densitometric analysis of the expression of related proteins. The results were shown as mean ± SD of three experiments with triplicate sets in each experiment. **p < 0.01 versus control, ## p < 0.01 versus treatment with glutamate alone in SH-SY5Y cells, ^^ p < 0.01 versus treatment with glutamate and GDNF in SH-SY5Y cells
Gas1 Silencing Promotes the Protective Effect of GDNF via Inhibition of Mitochondrial-Dependent Cell Apoptosis Signaling Pathway
One of the prominent events of cell apoptosis is mitochondrial dysfunction (Lemasters et al. 1999). Literature indicated that Gas1 could induce cell apoptosis through mediating mitochondrial dysfunction (Zarco et al. 2012). In this study, the expression of cytochrome c (Cyt-c) and activity of caspases were assessed. As shown in Figs. 6 and 7, glutamate treatment significantly down-regulated the expression of Cyt-c in mitochondria and up-regulated the activities of initiator caspase-9 and effector caspase-3; however, this effect was moderately reversed by GDNF co-treatment. Furthermore, Gas1 silencing significantly enhanced this effect of GDNF against mitochondrial dysfunction. Our results suggested Gas1 silencing promoted GDNF’s protective effect via inhibiting the mitochondrial-dependent cell apoptosis signaling pathway.

Gas1 silencing promotes the protective effect of GDNF against glutamate-induced cytotoxicity through modulating the apoptotic signaling pathways. SH-SY5Y cells with or without Gas1 knockdown were treated with glutamate (10 mM) in the absence or presence of GDNF (50 ng/mL) for 24 h. a The expression of cytochrome c in mitochondria (Cyt-c Mit) and in cytosol (Cyt-c Cyt) was assessed by Western blot analysis. b The densitometric analysis of the expression of Cyt-c Mit and Cyt-c Cyt. The results were shown as mean ± SD of three experiments with triplicate sets in each experiment. **p < 0.01 versus control, ## p < 0.01 versus treatment with glutamate alone in SH-SY5Y cells, ^^ p < 0.01 versus treatment with glutamate and GDNF in SH-SY5Y cells

The activities of caspase-3 and -9 were determined by fluorometric assay. SH-SY5Y cells with or without Gas1 knockdown were treated with glutamate (10 mM) in the absence or presence of GDNF (50 ng/mL) for 24 h and the activities of caspases were determined. The results were shown as mean ± SD of three experiments with triplicate sets in each experiment. **p < 0.01 versus control, ## p < 0.01 versus treatment with glutamate alone in SH-SY5Y cells, ^^ p < 0.01 versus treatment with glutamate and GDNF in SH-SY5Y cells
Discussion
GDNF, a distant member of the TGF-β family, is a key factor to maintain several cell populations in the CNS including dopaminergic and motor neurons. It also participates in the survival and differentiation of peripheral neurons such as enteric, sympathetic, and parasympathetic (Saarma 2000; Hansford and Marshall 2005). GDNF was found to have survival-promoting and restorative effects for midbrain dopaminergic neurons in vitro and in animal models of PD (Lin et al. 1993). This discovery raised great expectations to develop GDNF into a potent drug to cure or ameliorate the pathological symptoms of neurodegenerative disease such as PD. However, despite the positive outcome of small open-label clinical trials, other studies failed to demonstrate the prominent therapeutic efficacy of GDNF (Nutt et al. 2003; Patel et al. 2005; Sherer et al. 2006).
GDNF exerts it effect through a unique multicomponent receptor system consisting of glycosylphosphatidylinositol-anchored co-receptor (GFRα1-4) and RET tyrosine kinase (Treanor et al. 1996). Gas1, an apparently unrelated protein, has recently been found to exhibit structural homology to GFRα, which suggests the potential role of Gas1 in modulating the biological responses induced by GDNF. Gas1 is involved in cell growth arrest and able to induce apoptosis when over-expressed in different cell lines including those with neuronal and glial origins (Martinelli and Fan 2007). The potential effect of Gas1 is due to its capacity in inhibiting the GDNF-mediated survival pathway. Britt et al. reported that Gas1 could be induced in cultured corticohippocampal neurons committed to die after a brief exposure to NMDA, suggesting Gas1 is possibly a potent modulator in excitotoxin-induced cell injury (Mellstrom et al. 2002). The demonstration of previous reports provided us with a rational basis to test our hypothesis that Gas1 expression induced by excitotoxin in neuronal cells may interfere with the protective effect of GDNF.
Human SH-SY5Y neuroblastoma cells is a dopaminergic neuronal cell line which has been widely used as a suitable in vitro model for a chronic, progressive neuronal disease compared to primary neurons. In our study, SH-SY5Y cells were used to determine the expression profile of Gas1 with or without the treatment of glutamate (10 mM) (Maruyama et al. 2004). Our findings showed that treatment of glutamate induced a rapid accumulation of Gas1 protein in cells, which indicated Gas1 was likely involved in glutamate-induced neuronal death. It has been reported that GDNF could attenuate excitotoxin-induced neuronal death; however, this effect was not pronounced to be clinically important. In order to determine whether Gas1 silencing influenced the protective effect of GDNF, we performed experiments in the Gas1 knockdown cells. We found that Gas1 silencing could markedly enhance GDNF’s protective effect against glutamate-induced cytotoxicity as well as apoptosis in human SH-SY5Y neuroblastoma cells.
To further investigate the molecular mechanisms underlying the synergistic effect of Gas1 silencing and GDNF, we investigated the relevant survival and apoptotic signaling pathways. Previous reports demonstrated that Gas1 disrupted GDNF signaling through interfering the downstream Akt phosphorylation (Mellstrom et al. 2002). In our present study, GDNF induced the phosphorylation of Ret and thus activated Akt/PI3 K survival signaling pathway in SH-SY5Y cells, and this effect was more pronounced in Gas1 knockdown cells. Moreover, Akt could enhance the survival of cells by blocking the function of pro-apoptotic factors and processes (Gottlieb et al. 2002). For instance, Akt directly phosphorylates and inhibits the BH3-only protein Bad (Datta et al. 1997; Zundel and Giaccia 1998). The phosphorylation of S136 residue of Bad is important for the Akt-induced survival of neurons (Yu et al. 2005). In this study, we observed that the treatment of GDNF significantly attenuated the glutamate-induced dephosphorylation of Bad in the Gas1 knockdown cells, which effect was accompanied with the inhibited release of Cyt-c to the cytosol and the activation of caspases (caspase 3 and caspase 9). Our observation suggested that Gas1 silencing enhanced GDNF’s protective effect via activating PI3 K/Akt survival signaling pathway as well as inhibiting mitochondrial-dependent apoptotic signaling pathway via Bad dephosphorylation blockade.
In conclusion, these data extensively suggested the hypothesis that Gas1 silencing could significantly promote the neuroprotective effect of GDNF against glutamate-induced neurotoxicity in human SH-SY5Y neuroblastoma cells. Till now, there is limited progress of therapeutic development of GDNF in preventing and treating neurodegenerative diseases due to the unsatisfied outcome of clinical trials, but there is still reason to hope that trophic factor therapy may become a reality for patients with neurodegenerative diseases. The novel information gained in this study significantly contributes to elucidating the limited beneficial effect of GDNF in these clinical trials; however, this study is still at an early stage. Further in vivo studies are necessary to confirm the negative effect of Gas1 on GDNF therapy in the neurodegenerative diseases. Finally, our study may provide a novel therapeutic approach in the optimization of GDNF therapies targeting at neurodegenerative diseases.
Reference
Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P (1995) Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15:961–973
Beal MF (1992) Mechanisms of excitotoxicity in neurologic diseases. FASEB J 6:3338–3344
Bonde C, Kristensen BW, Blaabjerg M, Johansen TE, Zimmer J, Meyer M (2000) GDNF and neublastin protect against NMDA-induced excitotoxicity in hippocampal slice cultures. NeuroReport 11:4069–4073
Cabrera JR, Sanchez-Pulido L, Rojas AM, Valencia A, Mañes S, Naranjo JR, Mellström B (2006) Gas1 is related to the glial cell-derived neurotrophic factor family receptors α and regulates Ret signaling. J Biol Chem 281:14330–14339
Chalak LF, Rouse DJ (2011) Neuroprotective approaches: before and after delivery. Clin Perinatol 38:455–470
Choi-Lundberg DL, Lin Q, Chang Y-N, Chiang YL, Hay CM, Mohajeri H, Davidson BL, Bohn MC (1997) Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science 275:838–841
Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Del Sal G, Ruaro ME, Philipson L, Schneider C (1992) The growth arrest-specific gene, gas1, is involved in growth suppression. Cell 70:595–607
Edgar D, Barde Y-A (1983) Neuronal growth factors. Trends Neurosci 6:260–262
Evdokiou A, Cowled PA (1998) Tumor-suppressive activity of the growth arrest-specific gene GAS1 in human tumor cell lines. Int J Cancer 75:568–577
Ghribi O, Herman MM, Forbes MS, DeWitt DA, Savory J (2001) GDNF protects against aluminum-induced apoptosis in rabbits by upregulating Bcl-2 and Bcl-XL and inhibiting mitochondrial Bax translocation. Neurobiology of Disease 8:764–773
Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF (2010) Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta 1802:122–134
Gottlieb TM, Leal J, Seger R, Taya Y, Oren M (2002) Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene 21:1299–1303
Gratacòs E, Pérez-Navarro E, Tolosa E, Arenas E, Alberch J (2001) Neuroprotection of striatal neurons against kainate excitotoxicity by neurotrophins and GDNF family members. J Neurochem 78:1287–1296
Greenamyre JT, Porter R (1994) Anatomy and physiology of glutamate in the CNS. Neurology 44:S7–S13
Hansford LM, Marshall GM (2005) Glial cell line-derived neurotrophic factor (GDNF) family ligands reduce the sensitivity of neuroblastoma cells to pharmacologically induced cell death, growth arrest and differentiation. Neurosci Lett 389:77–82
Kruger NJ (1994) The Bradford method for protein quantitation. Methods Mol Biol 32:9–15
Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R, Brooks DJ, Hotton G, Moro E, Heywood P (2006) Randomized controlled trial of intraputamenal glial cell line–derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 59:459–466
Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, Nishimura Y, Nieminen A-L, Herman B (1999) Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr 31:305–319
Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F (1993) GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260:1130–1132
López-Ramírez MA, Domínguez-Monzón G, Vergara P, Segovia J (2008) Gas1 reduces Ret tyrosine 1062 phosphorylation and alters GDNF-mediated intracellular signaling. Int J Dev Neurosci 26:497–503
Martinelli DC, Fan C-M (2007) The role of Gas1 in embryonic development and its implications for human disease. Cell Cycle 6:2650–2655
Maruyama W, Nitta A, Shamoto-Nagai M, Hirata Y, Akao Y, Yodim M, Furukawa S, Nabeshima T, Naoi M (2004) N-Propargyl-1 (R)-aminoindan, rasagiline, increases glial cell line-derived neurotrophic factor (GDNF) in neuroblastoma SH-SY5Y cells through activation of NF-κB transcription factor. Neurochem Int 44:393–400
Mellstrom B, Cena V, Lamas M, Perales C, Gonzalez C, Naranjo JR (2002) Gas1 is induced during and participates in excitotoxic neuronal death. Mol Cell Neurosci 19:417–429
Nutt J, Burchiel K, Comella C, Jankovic J, Lang A, Laws E, Lozano A, Penn R, Simpson R, Stacy M (2003) Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 60:69–73
Patel NK, Bunnage M, Plaha P, Svendsen CN, Heywood P, Gill SS (2005) Intraputamenal infusion of glial cell line–derived neurotrophic factor in PD: a two-year outcome study. Ann Neurol 57:298–302
Pizza V, Agresta A, D’Acunto CW, Festa M, Capasso A (2011) Neuroinflamm-aging and neurodegenerative diseases: an overview. CNS Neurol Disord Drug Targets 10:621–634
Saarma M (2000) GDNF—a stranger in the TGF-β superfamily? Eur J Biochem 267:6968–6971
Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71:35–48
Schueler-Furman O, Glick E, Segovia J, Linial M (2006) Is GAS1 a co-receptor for the GDNF family of ligands? Trends Pharmacol Sci 27:72–77
Sherer TB, Fiske BK, Svendsen CN, Lang AE, Langston JW (2006) Crossroads in GDNF therapy for Parkinson’s disease. Mov Disord 21:136–141
Treanor JJ, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahashi M, Vandlen R, Henderson CE, Rosenthal A (1996) Characterization of a multicomponent receptor for GDNF. Nature 382:80–83
Ueda K, Fukui Y, Kageyama H (1994) Amyloid β protein-induced neuronal cell death: neurotoxic properties of aggregated amyloid β protein. Brain Res 639:240–244
van Meerloo J, Kaspers GJ, Cloos J (2011) Cell sensitivity assays: the MTT assay. Methods Mol Biol 731:237–245
Wang Y, Qin Z-H (2010) Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 15:1382–1402
Yan Q, Matheson C, Lopez OT (1995) In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature 373:341–344
Yu F, Sugawara T, Maier CM, Hsieh LB, Chan PH (2005) Akt/Bad signaling and motor neuron survival after spinal cord injury. Neurobiol Dis 20:491–499
Yuan J, Lipinski M, Degterev A (2003) Diversity in the mechanisms of neuronal cell death. Neuron 40:401–413
Zarco N, González-Ramírez R, González RO, Segovia J (2012) GAS1 induces cell death through an intrinsic apoptotic pathway. Apoptosis 17:627–635
Zarco N, Bautista E, Cuellar M, Vergara P, Flores-Rodriguez P, Aguilar-Roblero R, Segovia J (2013) Growth arrest specific 1 (GAS1) is abundantly expressed in the adult mouse central nervous system. J Histochem Cytochem 61:731–748
Zhu X, Wang K, Zhang K, Zhu L, Zhou F (2014) Ziyuglycoside II induces cell cycle arrest and apoptosis through activation of ROS/JNK pathway in human breast cancer cells. Toxicol Lett 227:65–73
Zundel W, Giaccia A (1998) Inhibition of the anti-apoptotic PI (3) K/Akt/Bad pathway by stress. Genes Dev 12:1941–1946
Acknowledgments
This work was supported by grants from the National Natural Science Foundation (81300787), the Natural Science Foundation of Jiangsu Province (BK2011168, BK2012105, BK20141103), and the Major Project of Wuxi Municipal Health Bureau (ZS201401).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, K., Zhu, X., Zhang, K. et al. Gas1 Knockdown Increases the Neuroprotective Effect of Glial Cell-Derived Neurotrophic Factor Against Glutamate-Induced Cell Injury in Human SH-SY5Y Neuroblastoma Cells. Cell Mol Neurobiol 36, 603–611 (2016). https://doi.org/10.1007/s10571-015-0241-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-015-0241-3