
Abstract
Accumulated evidence has shown that excessive reactive oxygen species (ROS) have been implicated in neuronal cell death related with various chronic neurodegenerative disorders. This study was designed to explore neuroprotective effects of ethyl acetate extract of Arctium lappa L. roots (EAL) on hydrogen peroxide (H2O2)-induced cell injury in human SH-SY5Y neuroblastoma cells. The cell viability was significantly decreased after exposure to 200 μM H2O2, whereas pretreatment with different concentrations of EAL attenuated the H2O2-induced cytotoxicity. Hoechst 33342 staining indicated that EAL reversed nuclear condensation in H2O2-treated cells. Meanwhile, TUNEL assay with DAPI staining showed that EAL attenuated apoptosis was induced by H2O2. Pretreatment with EAL also markedly elevated activities of antioxidant enzyme (GSH-Px and SOD), reduced lipid peroxidation (MDA) production, prevented ROS formation, and the decrease of mitochondrial membrane potential. In addition, EAL showed strong radical scavenging ability in 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) assays. Furthermore, EAL inhibited H2O2-induced apoptosis by increases in the Bcl-2/Bax ratio, decreases in cytochrome c release, and attenuation of caspase-3, caspase-9 activities, and expressions. These findings suggest that EAL may be regarded as a potential antioxidant agent and possess potent neuroprotective activity against H2O2-induced injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reactive oxygen species (ROS) are essential signaling molecules in aerobic respiration process, since they regulate many metabolic reactions (Jomova and Valko 2011). However, excessively high levels of ROS may induce severe cell damage. ROS can oxidize essential biological macromolecules, such as membrane lipids, DNAs, and proteins, thereby activating several signaling pathways that lead to cellular damage and death (Choi et al. 2014; Park et al. 2010). In addition, accumulated evidences have shown that excessive ROS have been implicated in neuronal cell death related with various chronic neurodegenerative disorders, such as multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease (Yan et al. 2013). Therefore, the inhibition of ROS generation could prove beneficial in the prevention and treatment of these diseases.
Hydrogen peroxide (H2O2) is a major contributor to ROS. H2O2 is generated as a by-product of enzymatic action during the processes of dopamine oxidation, amyloid aggregation, and brain ischemia reperfusion (Nelson et al. 2005). Then, H2O2 is readily converted into highly reactive hydroxyl radicals by the Fenton reaction and can attack biological molecules. Further, this oxidative damage could lead to decreases in antioxidant enzyme activities, mitochondrial dysfunction, and apoptosis in neuronal cells. However, these effects can be blocked by the addition of antioxidants (Ghaffari et al. 2014). Many synthetic chemicals have the strong ability to scavenge radicals, but are usually accompanied by many adverse effects. Recently, great attentions have been given to natural products with neuroprotective potential. These natural substances act as radical scavengers to protect cells from oxidative damage (Ju et al. 2012; Si et al. 2013).
The root of Arctium lappa Linne, known as burdock or bardana, is a very popular cultivated edible vegetable in many countries. Recently, various studies have demonstrated the biological properties of this species, including antiallergic (Sohn et al. 2011), antibacterial against gram-positive and gram-negative bacteria (Gentil et al. 2006), and antiulcerogenic (de Almeida et al. 2012) activities. Further, lignans from A. lappa and their ability to inhibit lipopolysaccharide (LPS)-stimulated nitric oxide production and pro-inflammatory cytokines secretion were described (Hyam et al. 2013; Park et al. 2007). In addition, our group recently reported that the neuroprotective effects of the ethyl acetate extract of A. lappa roots (EAL) against glutamate-mediated cytotoxicity in PC12 cells (Tian et al. 2014). The total phenolic content (mg gallic acid/g extract) was determined as 88.90 ± 1.63 mg/g in the EAL. However, whether EAL displayed antioxidant activities and protective effects on another neuronal injury model, H2O2-induced cell injury were unknown.
In continuation, the present study aimed to investigate the antioxidant activities and neuroprotective potential of EAL on H2O2-induced cell injury and explored its potential mechanisms in SH-SY5Y cells.
Materials and Methods
Materials
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 0.25 % trypsin–EDTA, and dimethyl sulfoxide (DMSO) were purchased from Amresco (Solon, OH, USA). The kit for lactate dehydrogenase (LDH) assay was obtained from Nanjing Jiancheng (Nanjing, China). The 2,7′-dichlorofluorescin diacetate (DCF-DA), the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) apoptosis assay kit, 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI), caspase-3 and caspase-9 activity kits, superoxide dismutase (SOD), malondialdehyde (MDA), glutathione peroxidase (GSH-Px), and total antioxidant capacity assay kits were all acquired from Beyotime (Haimen, China). Rhodamine 123, Hoechst 33342, and H2O2 were obtained from Sigma-Chemical (St. Louis, MO, USA). We purchased fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM), and F12 medium from Hyclone (Logan, UT, USA). The following antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA): rabbit anti-Bax, rabbit anti-Bcl-2, rabbit anti-caspase-3, rabbit anti-caspase-9, mouse anti-cytochrome c, and goat secondary antibodies. All other reagents and chemicals were of analytical grade.
Preparation of EAL
The preparation of EAL was performed as described in our previous study (Tian et al. 2014). Briefly, a voucher specimen (TX2012NBG) of A. lappa roots was deposited at the Department of Pharmacy, General Hospital of Shenyang Military Area Command, Shenyang, China. 20 g of powder was extracted two times with 55 % ethanol. The crude extract (5.34 g, 26.70 %) was then partitioned with two times volume amounts of ethyl acetate for three times to obtain EAL (0.2380 g, 4.46 %).
Cell Culture
Human neuroblastoma SH-SY5Y cells were cultured in DMEM-F12 (1:1) medium supplemented with 10 % FBS. Cells were incubated at 37 °C in a humidified atmosphere incubator of 5 % CO2. The culture medium was changed every other day, and the cells were subcultured once they reached 70–80 % confluence.
Cell Viability Assay
Cytoprotective activity of EAL on H2O2-induced cell injury was assessed by MTT assay. Briefly, SH-SY5Y cells were seeded into 96-well plates at a density of 1 × 104 cells/well and grown in the incubator for 24 h. H2O2 was prepared prior to each experiment. Cells pretreated for 12 h with different concentrations of EAL (20, 40, and 80 μg/mL) were exposed to 200 μM H2O2 for 3 h. Then 20 μL of the MTT solution (5 mg/mL) was added into each well. After incubation for 4 h at 37 °C, the medium was slowly removed, and 150 μL of DMSO was added. The absorbance was measured at 490 nm with a microplate reader (ELX 800, Bio-tek, USA). The value for cell viability was expressed as the percentage of the control value.
LDH Assay
SH-SY5Y cells were seeded into 96-well culture plates at a density of 1 × 104 cells/well. The cells were incubated with 200 μM H2O2 for 3 h with or without the pretreatment of EAL. Then, the supernatant was used in the LDH assay according to the manufacturer’s instructions. The LDH activity was measured by monitoring the reduction of pyruvic acid. The absorbance of each sample was determined at 450 nm with a microplate reader. LDH leakage was calculated as the percentage of the control group.
Hoechst 33342 Staining
SH-SY5Y cells were seeded into 6-well plates at a density of 2 × 105 cells/well. The cells were exposed to 200 μM H2O2 for 3 h with or without EAL pretreatment. After the treatment, the cells were washed with PBS solution and loaded with 10 μg/mL Hoechst 33342 dye for 15 min (Tan et al. 2013). Then, the cells were visualized under a fluorescence microscope (IX71, Olympus, Japan), and cell images were recorded.
TUNEL Assay
The cells treated as indicated were washed with PBS twice and subsequently fixed with 4 % paraformaldehyde for 30 min. After washing with PBS once again, the cells were permeabilized with 0.1 % Triton X-100 for 5 min on ice and rinsed with PBS twice. Then they were incubated with TUNEL staining solution for 1 h at 37 °C. After washing with PBS twice, the cells were stained with DAPI for 15 min. Subsequently, the cells were observed by a fluorescence microscope (IX71, Olympus, Japan). Fifteen random fields were selected for each experimental condition.
Measurement of Cellular Antioxidant Enzymes
Following the treatment as described above, SH-SY5Y cells were washed twice with ice-cold PBS and collected. The solution was centrifuged at 2,000 rpm for 10 min. Then cell pellets were lysed by the cell lysis buffer. After centrifugation at 12,000×g for 10 min at 4 °C, the supernatant was used in the activity assays of GSH-Px and SOD according to the manufacturer’s instructions. The activities of GSH-Px and SOD were measured at 340 and 450 nm, respectively.
Lipid Peroxidation Assay
Lipid peroxidation was determined by the thiobarbituric acid (TBA) assay. Briefly, the 100 μL lysates and 200 μL MDA working solution were pipetted into 0.5 mL tubes and incubated for 15 min. After centrifugation at 1,000×g for 10 min, the absorbance of the supernatant was measured at 532 nm (Qian et al. 2010). The levels of MDA were expressed as nmol/mg of protein.
Total Antioxidant Capacity Assay
The total antioxidant capacity was measured by a rapid 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) assay kit according to the manufacturer’s instructions. The absorbance of each sample was determined by the ability to inhibit the formation of ABTS at 414 nm. ABTS radical scavenging rate was calculated as follows: ABTS scavenging rate = (Acontrol − Asample)/Acontrol × 100 %. The IC50 value that the concentration of EAL required to scavenge 50 % of ABTS radical was also calculated.
Measurement of Intracellular ROS Levels
The intracellular ROS level was assessed by using the DCFH-DA fluorescent probe. SH-SY5Y cells (2 × 105 cells/well) were incubated in 6-well plates for 24 h, and then treated with H2O2 for 3 h in the presence or absence of EAL. The cells were washed twice with PBS and loaded with DCFH-DA (final concentration 5 μM/L) in serum-free medium (Menazza et al. 2010). After incubation for 30 min at 37 °C, the cells were observed at 525 nm with a fluorescence microscope (IX71, Olympus, Japan).
Determination of Mitochondrial Membrane Potential (MMP)
The fluorescent Rhodamine 123 dye was used for measuring MMP (Luo et al. 2011). A decrease in fluorescence intensity represents mitochondrial membrane depolarization. Following the treatment as described above, the cells were rinsed twice with PBS and then incubated with Rhodamine 123 (final concentration 1 mM/L) for 30 min at room temperature. After these treatments, the images of the stained cells were observed using a fluorescence microscope (IX71, Olympus, Japan).
Caspase-3 and Caspase-9 Activities Assay
Caspase-3 and caspase-9 activities were measured by commercial assay kits following the manufacturer’s protocols. Briefly, after H2O2 exposure, the collected cells were washed with ice-cold PBS, and incubated with the cell lysis buffer on ice for 30 min. Then the cell lysates were centrifuged at 12,000×g for 10 min at 4 °C to obtain the supernatants. A reaction mixture containing 70 μL of reaction buffer, 20 μL of the supernatant, and 10 μL of caspase-3 (Ac-DEVD-pNA) or caspase-9 substrate (Ac-LEHD-pNA) was added into 96-well plates, respectively. After incubation at 37 °C for 1 h, the absorbance of yellow pNA was determined at 405 nm. The enzyme activities were standardized to milligram protein.
Western Blot Analysis
At the end of treatment, the cells were harvested, washed twice with ice-cold PBS, and lysed with the ice-cold lysis buffer for 30 min. Then, the lysed cells were centrifuged at 12,000×g for 15 min. The supernatant was collected, and the protein content was measured by a BCA Protein Assay Kit, followed by electrophoretic separation on a 12 % SDS polyacrylamide gel. After transferring to PVDF membranes, all protein samples were incubated overnight with primary antibodies against Bax (1:800), Bcl-2 (1:500), cytochrome c (1:600), caspase-3 (1:500), caspase-9 (1:800), and β-actin (1:1000). Then the blots were reacted with horseradish peroxidase-conjugated secondary antibodies (1:5000) for 2 h at room temperature. Antibody-bound proteins were detected by an enhanced chemiluminescence (ECL) method. The β-actin protein served as an internal control in western blot analysis.
Statistical Analysis
All experiments were carried out in triplicate. The data were expressed as mean ± standard deviation (SD). Significant differences were calculated in SPSS 16.0 with one-way analysis of variance (ANOVA), followed by the post hoc LSD test. p values less than 0.05 were considered to be significant.
Results
Effects of EAL on H2O2-Induced Cytotoxicity in SH-SY5Y Cells
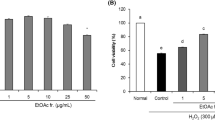
In our preliminary experiments, treatment of SH-SY5Y cells with various concentrations of EAL (20–500 μg/mL) alone did not show any significant effects on the cells proliferation over 24 h (data not shown). After exposure to H2O2, dose-dependent decreases in cell viabilities were investigated by MTT assay. The cell viability was markedly decreased when SH-SY5Y cells were treated with H2O2 for 3 h, and the IC50 value of H2O2 was 234.67 μmol/L. Therefore, 200 μM H2O2 was chosen to induce injury for the subsequent experiments.
The cells that exposed to 200 μM H2O2 for 3 h showed a remarkable decrease in cell viability (62.01 ± 3.50 %, compared with the control group). However, pretreatment with different concentrations of EAL (20, 40, and 80 μg/mL) restored the cell viability in a dose-dependent manner. The cell viability was significantly increased to 68.11 ± 2.16 %, 76.35 ± 1.51 %, and 85.17 ± 2.52 % as compared with the control group, respectively (Fig. 1a).

Effects of EAL on H2O2-induced cytotoxicity in SH-SY5Y cells. a The cell viability was measured by MTT assay. b The release of LDH into extracellular medium was assessed by using the LDH assay. SH-SY5Y cells were pretreated with EAL for 12 h (20, 40, and 80 μg/mL) prior to 200 μM H2O2 for 3 h. Data were shown as mean ± SD (n = 4). ## p < 0.01 versus the control group. *p < 0.05 and **p < 0.01 versus the H2O2-treated group
To further investigate the protective effects provided by EAL against H2O2-induced cytotoxicity, the amount of the LDH released into the culture medium was measured. After cells were exposed to 200 μM H2O2, LDH leakage was increased to 814.46 ± 41.02 % of the control value, but this was markedly reduced to 530.27 ± 41.65 %, 382.17 ± 27.20 %, and 265.93 ± 19.99 % by EAL pretreatment at 20, 40, and 80 μg/mL, respectively (Fig. 1b).
Effects of EAL on H2O2-Induced Apoptosis in SH-SY5Y Cells
The control SH-SY5Y cells grew well and had round cell bodies under phase-contrast microscopy (Fig. 2a). However, incubation with 200 μM H2O2 for 3 h led to decreases in the number of viable cells, shrinkage, and aggregation of cell bodies. In contrast, pretreatment with EAL significantly attenuated the morphological changes of cell damage.

Effects of EAL on cell morphological changes induced by H2O2 in SH-SY5Y cells. a Morphological changes of SH-SY5Y cells were observed under a phase-contrast microscopy. b Nuclear morphological changes of the cells were observed using a fluorescence microscopy after Hoechst 33342 staining. Cells were pretreated with EAL at the indicated concentration for 12 h and then treated with 200 μM H2O2 for 3 h
Furthermore, Hoechst 33342 staining and TUNEL assay were performed to investigate whether EAL inhibited H2O2-induced apoptosis. As shown in Fig. 2b, nuclear condensation was presented in cells treated with H2O2. Meanwhile, TUNEL assay with DAPI staining indicated that DNA fragmentation was detected after H2O2 exposure (Fig. 3). However, pretreatment with EAL inhibited these characteristics of apoptosis.

Effects of EAL on H2O2-induced apoptosis in SH-SY5Y cells. a Representative photographs of fluorescent staining for DAPI (blue) and TUNEL (green). b Quantification analysis of TUNEL-positive cells. Cells were incubated with indicated concentrations of EAL for 12 h before exposure to 200 μM H2O2 for 3 h. Data are shown as mean ± SD (n = 3). ## p < 0.01 versus the control group. *p < 0.05 and **p < 0.01 versus the H2O2-treated group (Color figure online)
Effects of EAL on the Activities of GSH-Px and SOD, and MDA Production
Exposure of the cells to 200 μM H2O2 reduced the activities of GSH-Px and SOD to 55.03 % and 50.09 % of the control values, respectively (Fig. 4a, b). However, these effects were significantly reversed by pretreatment with EAL. Further, after incubation of SH-SY5Y cells with 200 μM H2O2, the MDA level was markedly increased to 219.43 % relative to the control group, whereas EAL reduced the MDA production in a dose-dependent manner (Fig. 4c).

Effects of EAL on (a) GSH-Px activities, (b) SOD activities and (c) MDA levels in H2O2-treated cells. Cells were incubated with different concentrations of EAL for 12 h before exposed to 200 μM H2O2 for 3 h. Data are shown as mean ± SD (n = 3). ## p < 0.01 versus the control group. *p < 0.05 and **p < 0.01 versus the H2O2-treated group. d Radical scavenging activity of EAL by ABTS assay. Inhibition is presented as percent inhibitions of ABTS radical cations compared with the control value
Radical Scavenging Activity of EAL
The radical scavenging activity of EAL with the ABTS method is shown in Fig. 4d. The inhibition rates of ABTS radical cations by EAL were 8.31 ± 1.48 %, 17.76 ± 1.55 %, 41.12 ± 1.80 %, and 80.72 ± 1.22 % at 20, 40, 80, and 160 μg/mL, respectively. In addition, the IC50 value for EAL in the ABTS antioxidant capacity assay was 83.48 μg/mL.
Effects of EAL on Intracellular ROS Production
As shown in Fig. 5, when the cells were exposed to 200 μM H2O2 alone, the intracellular ROS level was 196.7 ± 14.92 % of the control group. However, pretreatment with increasing concentrations of EAL at 20, 40, and 80 μg/mL significantly reduced the ROS production to 173.51 ± 10.85 %, 137.45 ± 9.86 %, and 121.54 ± 8.22 %, respectively.

Effects of EAL on H2O2-induced intracellular ROS production in SH-SY5Y cells. Cells were pretreated with different concentrations of EAL for 12 h and then exposed to 200 μM H2O2 for 3 h. ROS production was assessed with DCFH-DA fluorescence dye. a Control. b 200 μM H2O2. c 200 μM H2O2 + 20 μg/mL EAL. d 200 μM H2O2 + 40 μg/mL EAL. e 200 μM H2O2 + 80 μg/mL EAL. f The quantitative analysis of the mean DCF fluorescent intensity. Fifteen random fields of immunostained SH-SY5Y cells were chosen by using a ×40 objective. ## p < 0.01 versus the control group. *p < 0.05 and **p < 0.01 versus the H2O2-treated group (n = 3)
Effects of EAL on MMP
As illustrated in Fig. 6, the SH-SY5Y cells that treated with 200 μM H2O2 showed a significant decrease in MMP, 62.41 ± 4.59 % of the control group. In contrast, the fluorescence intensity of MMP was increased to 69.80 ± 5.29 %, 76.37 ± 4.12 %, and 83.82 ± 5.16 % by EAL at 20, 40, and 80 μg/mL, respectively.

Effects of EAL on H2O2-induced MMP loss in SH-SY5Y cells. After treated with EAL for 12 h, SH-SY5Y cells were exposed to 200 μM H2O2 for 3 h. MMP was determined by Rhodamine 123 fluorescence dye. a Control. b 200 μM H2O2. c 200 μM H2O2 + 20 μg/mL EAL. d 200 μM H2O2 + 40 μg/mL EAL. e 200 μM H2O2 + 80 μg/mL EAL. f The quantitative analysis of the mean rhodamine 123 fluorescent intensity. ## p < 0.01 versus the control group. *p < 0.05 and **p < 0.01 versus the H2O2-treated group (n = 3)
Effects of EAL on Caspase-3 and Caspase-9 Activities
After exposure to 200 μM H2O2, the activity of caspase-3 was significantly increased to about 4.21-fold higher than the control group. In contrast, EAL pretreatment inhibited caspase-3 activity by 25.37, 49.43, and 62.27 % at 20, 40, and 80 μg/mL, respectively. Caspase-9 activity showed the similar trend (Fig. 7).

Effects of EAL on enzymatic activities of a caspase-3 and b caspase-9. Cells were pretreated with different concentrations of EAL for 12 h prior to 200 μM H2O2 for 3 h. ## p < 0.01 versus the control group. *p < 0.05 and **p < 0.01 versus the H2O2-treated group (n = 3)
Effects of EAL on Protein Expressions
The Bax/Bcl-2 ratio was markedly increased to 602.94 ± 33.26 % of the control group after incubation of SH-SY5Y cells with 200 μM H2O2. Meanwhile, the release of cytochrome c was remarkably increased to 342.5 ± 19.03 % of the control value (Fig. 8). However, EAL significantly suppressed these trends.

Effects of EAL on protein expressions in H2O2-treated SH-SY5Y cells. After treated with EAL for 12 h, SH-SY5Y cells were incubated with 200 μM H2O2 for 3 h. a Bax, Bcl-2, b cytochrome c, c caspase-3 and d caspase-9 proteins were detected by western blot analysis. Data are shown as mean ± SD (n = 3). ## p < 0.01 versus the control group. *p < 0.05 and **p < 0.01 versus the H2O2-treated group
Also, treatment of SH-SY5Y cells with 200 μM H2O2 markedly induced the up-regulation of caspase-3 to 275.82 ± 22.12 % of the control group. In contrast, the up-regulation of caspase-3 was significantly inhibited by 20, 40, and 80 μg/mL EAL to 237.53 ± 16.75 %, 190.55 ± 13.33 %, and 168.57 ± 11.67 % of the control value, respectively. Similar results were observed in caspase-9 protein expression.
Discussion
Considerable efforts have been made in the search for natural substances with neuroprotective activities (Wang et al. 2014). Excessive H2O2 plays an important role in neuronal damage and subsequent cell death, while antioxidants attenuate oxidative stress by neutralizing free radicals such as ROS (Kumar and Khanum 2013). Therefore, H2O2-induced cytotoxicity is the common model applied to the investigation of neuroprotective antioxidants. In the present study, pretreatment of EAL inhibited decreases in cell viability, LDH release, and morphological changes in H2O2-treated cells. Considering the protective effect exerted by EAL on H2O2-induced cytotoxicity, EAL may be of value in neurodegenerative diseases and its underlying mechanisms need further investigation.
Exposure to free radicals is usually unavoidable for organisms in an aerobic environment. Normally, cells possess their own antioxidant defense system, mainly comprised of three major antioxidant enzymes, catalase (CAT), GPx and SOD, and a number of small non-enzyme molecules, such as oxidized and reduced glutathione(GSH) (Valko et al. 2007). By using reduced GSH, GPx catalyzes the reduction of peroxide to form oxidized GSH (GSSG) and water. In addition, SOD possesses the ability to catalyze the conversion of O2 − to hydrogen peroxide, while CAT converts hydrogen peroxide to water and oxygen. However, in H2O2-treated cells, H2O2 causes an imbalance between antioxidants defense system and the production of ROS, in favor of the later and leading to cellular disruption. This phenomenon is consistent with previous studies (Azmi et al. 2013; Guizani et al. 2011). In EAL treated cells, reversal of these effects suggests that EAL attenuated H2O2-induced oxidative damage via decreases in intracellular lipid peroxidation levels and increases in the activities of intracellular antioxidant enzymes. Moreover, earlier studies have indicated that H2O2 also decreases protein and mRNA levels of GSH-Px and SOD (Choi et al. 2014; Kumar and Khanum 2013). However, whether EAL has effects on protein and mRNA levels of these antioxidant enzymes need further research.
Oxidative stress has been observed to be involved in the development of mitochondrial dysfunctions, as contributing to open the mitochondrial permeability transition pore, resulting in depolarization of the MMP (Beal 2005; Venuprasad et al. 2013). Further, decreases in the integrity of the mitochondria could cause decreases in adenosine triphosphate production and increases in ROS production (Somayajulu et al. 2005). In addition, sustained elevated ROS levels may in turn impair mitochondrial function, and over-activate phospholipases and proteases, leading to irreversible membrane, chromatin, and organelle damage and ultimately cell death (Dumont and Beal 2011). We found that MMP was decreased in H2O2-treated cells, while EAL pretreatment reduced mitochondrial membrane depolarization. Therefore, we hypothesized that the ability of EAL to inhibit ROS formation may attribute to the preservation of MMP.
Previous study has shown that mitochondria-dependent apoptotic pathways are associated with H2O2-induced cytotoxicity in SH-SY5Y cells (Kwon et al. 2011). The antiapoptotic factor Bcl-2 that located in the outer mitochondrial membrane promotes cell survival. During the initiation phase of apoptosis, the pro-apoptotic factor Bax translocates from cytoplasm to the mitochondria and increases mitochondrial membrane permeability. Moreover, increased mitochondrial permeability can trigger the release of cytochrome c from the mitochondria to the cytosol, leading to activation of caspase-9, and then activation of caspase-3. Caspases are the family of cysteine proteases that are responsible for modulating cell death. Among them, caspase-3 is one of the main apoptotic executors and plays an essential role in neuronal apoptosis (Le et al. 2002). And caspase-9 is a major activator involved in mitochondrial-mediated apoptotic pathways attributing to caspase-3 activation. Following exposure to H2O2, the activated caspase-3 could stimulate DNA fragmentation factor, in turn activating endonucleases to cleave chromatin and eventually leads to cell death (Han et al. 2002; Kim et al. 2011). In addition, enzymatic activities of caspase-3 and caspase-9 were up-regulated by H2O2 treatment. However, these effects were reversed by EAL pretreatment. Therefore, our data indicate that the inhibition of cytochrome c release and apoptosis-related proteins activation may contribute to protection of EAL against H2O2-induced apoptosis.
Conclusion
In conclusion, the present study suggests that EAL can provide protection against H2O2-induced cell death by its antioxidant and antiapoptotic activities in SH-SY5Y cells. The protective effects of EAL may be mediated by the recovery of antioxidant enzymes activities, the reduction of lipid peroxidation, and ROS generation. Moreover, neuroprotective mechanisms of EAL may be associated with apoptosis inhibition by increases in the Bcl-2/Bax ratio, decreases in cytochrome c release, and attenuation of caspase-3 and caspase-9 activities and expressions. Together, these data suggest that EAL may be a potential neuroprotective source for treating neurodegenerative disorders such as Parkinson’s and Alzheimer’s diseases.
References
Azmi NH, Ismail N, Imam MU, Ismail M (2013) Ethyl acetate extract of germinated brown rice attenuates hydrogen peroxide-induced oxidative stress in human SH-SY5Y neuroblastoma cells: role of anti-apoptotic, pro-survival and antioxidant genes. BMC Complement Altern Med 13:177. doi:10.1186/1472-6882-13-177
Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58:495–505. doi:10.1002/ana.20624
Choi DJ, Kim SL, Choi JW, Park YI (2014) Neuroprotective effects of corn silk maysin via inhibition of H2O2-induced apoptotic cell death in SK-N-MC cells. Life Sci 109:57–64. doi:10.1016/j.lfs.2014.05.020
de Almeida ABA, Luiz-Ferreira A, Cola M, Magri LD, Batista LM, de Paiva JA, Trigo JR, Souza-Brito ARM (2012) Anti-ulcerogenic mechanisms of the sesquiterpene lactone onopordopicrin-enriched fraction from Arctium lappa L. (Asteraceae): role of somatostatin, gastrin, and endogenous sulfhydryls and nitric oxide. J Med Food 15:378–383. doi:10.1089/jmf.2011.0025
Dumont M, Beal MF (2011) Neuroprotective strategies involving ROS in Alzheimer disease. Free Radical Bio Med 51:1014–1026. doi:10.1016/j.freeradbiomed.2010.11.026
Gentil M, Pereira JV, Sousa YTCS, Pietro R, Neto MDS, Vansan LP, de Castro França S (2006) In vitro evaluation of the antibacterial activity of Arctium lappa as a phytotherapeutic agent used in intracanal dressings. Phytother Res 20:184–186. doi:10.1002/ptr.1829
Ghaffari H, Ghassam B, Chandra Nayaka S, Ramachandra Kini K, Prakash HS (2014) Antioxidant and neuroprotective activities of Hyptis suaveolens (L.) Poit. against oxidative stress-induced neurotoxicity. Cell Mol Neurobiol 34:323–331. doi:10.1007/s10571-013-0016-7
Guizani N, Waly MI, Ali A, Al-Saidi G, Singh V, Bhatt N, Rahman MS (2011) Papaya epicarp extract protects against hydrogen peroxide-induced oxidative stress in human SH-SY5Y neuronal cells. Exp Biol Med (Maywood) 236:1205–1210. doi:10.1258/ebm.2011.011031
Han BH, Xu D, Choi J, Han Y, Xanthoudakis S, Roy S, Tam J, Vaillancourt J, Colucci J, Siman R, Giroux A, Robertson GS, Zamboni R, Nicholson DW, Holtzman DM (2002) Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury. J Biol Chem 277:30128–30136. doi:10.1074/jbc.M202931200
Hyam SR et al (2013) Arctigenin ameliorates inflammation in vitro and in vivo by inhibiting the PI3 K/AKT pathway and polarizing M1 macrophages to M2-like macrophages. Eur J Pharmacol 708:21–29. doi:10.1016/j.ejphar.2013.01.014
Jomova K, Valko M (2011) Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharm Des 17:3460–3473. doi:10.2174/138161211798072463
Ju H-Y, Chen SC, Wu K-J, Kuo H-C, Hseu Y-C, Ching H, Wu C-R (2012) Antioxidant phenolic profile from ethyl acetate fraction of Fructus Ligustri Lucidi with protection against hydrogen peroxide-induced oxidative damage in SH-SY5Y cells. Food Chem Toxicol 50:492–502. doi:10.1016/j.fct.2011.11.036
Kim S, Park SE, Sapkota K, Kim MK, Kim SJ (2011) Leaf extract of Rhus verniciflua Stokes protects dopaminergic neuronal cells in a rotenone model of Parkinson’s disease. J Pharm Pharmacol 63:1358–1367. doi:10.1111/j.2042-7158.2011.01342.x
Kumar KH, Khanum F (2013) Hydroalcoholic extract of Cyperus rotundus ameliorates H2O2-induced human neuronal cell damage via its anti-oxidative and anti-apoptotic machinery. Cell Mol Neurobiol 33:5–17. doi:10.1007/s10571-012-9865-8
Kwon S-H, Kim J-A, Hong S-I, Jung Y-H, Kim H-C, Lee S-Y, Jang C-G (2011) Loganin protects against hydrogen peroxide-induced apoptosis by inhibiting phosphorylation of JNK, p38, and ERK 1/2 MAPKs in SH-SY5Y cells. Neurochem Int 58:533–541. doi:10.1016/j.neuint.2011.01.012
Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA, Moskowitz MA (2002) Caspase activation and neuroprotection in caspase-3- deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A 99:15188–15193. doi:10.1073/pnas.232473399
Luo T, Zhang H, Zhang WW, Huang JT, Song EL, Chen SG, He F, Xu J, Wang HQ (2011) Neuroprotective effect of jatrorrhizine on hydrogen peroxide-induced cell injury and its potential mechanisms in PC12 cells. Neurosci Lett 498:227–231. doi:10.1016/j.neulet.2011.05.017
Menazza S, Blaauw B, Tiepolo T, Toniolo L, Braghetta P, Spolaore B, Reggiani C, Di Lisa F, Bonaldo P, Canton M (2010) Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum Mol Genet 19:4207–4215. doi:10.1093/hmg/ddq339
Nelson SK, Bose S, Rizeq M, McCord JM (2005) Oxidative stress in organ preservation: a multifaceted approach to cardioplegia. Biomed Pharmacother 59:149–157. doi:10.1016/j.biopha.2005.03.007
Park SY, Hong SS, Han XH, Hwang JS, Lee D, Ro JS, Hwang BY (2007) Lignans from Arctium lappa and their inhibition of LPS-induced nitric oxide production. Chem Pharm Bull (Tokyo) 55:150–152. doi:10.1248/cpb.55.150
Park SE, Kim S, Sapkota K, Kim SJ (2010) Neuroprotective effect of Rosmarinus officinalis extract on human dopaminergic cell line, SH-SY5Y. Cell Mol Neurobiol 30:759–767. doi:10.1007/s10571-010-9502-3
Qian J, Jiang F, Wang B, Yu Y, Zhang X, Yin Z, Liu C (2010) Ophiopogonin D prevents H2O2-induced injury in primary human umbilical vein endothelial cells. J Ethnopharmacol 128:438–445. doi:10.1016/j.jep.2010.01.031
Si CL, Shen T, Jiang YY, Wu L, Yu GJ, Ren XD, Xu GH, Hu WC (2013) Antioxidant properties and neuroprotective effects of isocampneoside II on hydrogen peroxide-induced oxidative injury in PC12 cells. Food Chem Toxicol 59:145–152. doi:10.1016/j.fct.2013.05.051
Sohn EH, Jang SA, Joo H, Park S, Kang SC, Lee CH, Kim SY (2011) Anti-allergic and anti-inflammatory effects of butanol extract from Arctium Lappa L. Clin Mol Allergy 9:4. doi:10.1186/1476-7961-9-4
Somayajulu M, McCarthy S, Hung M, Sikorska M, Borowy-Borowski H, Pandey S (2005) Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by coenzyme Q10. Neurobiol Dis 18:618–627. doi:10.1016/j.nbd.2004.10.021
Tan JW, Tham CL, Israf DA, Lee SH, Kim MK (2013) Neuroprotective effects of biochanin A against glutamate-induced cytotoxicity in PC12 cells via apoptosis inhibition. Neurochem Res 38:512–518. doi:10.1007/s11064-012-0943-6
Tian X, Sui S, Huang J, Bai J-P, Ren T-S, Zhao Q-C (2014) Neuroprotective effects of Arctium lappa L. roots against glutamate-induced oxidative stress by inhibiting phosphorylation of p38, JNK and ERK 1/2 MAPKs in PC12 cells. Environ Toxicol Phar 38:189–198. doi:10.1016/j.etap.2014.05.017
Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell B 39:44–84. doi:10.1016/j.biocel.2006.07.001
Venuprasad MP, Hemanth Kumar K, Khanum F (2013) Neuroprotective effects of hydroalcoholic extract of Ocimum sanctum against H2O2 induced neuronal cell damage in SH-SY5Y cells via its antioxidative defence mechanism. Neurochem Res 38:2190–2200. doi:10.1007/s11064-013-1128-7
Wang K, Zhu L, Zhu X, Zhang K, Huang B, Zhang J, Zhang Y, Zhu L, Zhou B, Zhou F (2014) Protective effect of paeoniflorin on Aβ25–35-induced SH-SY5Y cell injury by preventing mitochondrial dysfunction. Cell Mol Neurobiol 34:227–234. doi:10.1007/s10571-013-0006-9
Yan MH, Wang X, Zhu X (2013) Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62:90–101. doi:10.1016/j.freeradbiomed.2012.11.014
Acknowledgments
This project was supported by the Key National Science & Technology Specific Project of China (2014ZX09J14101-05C).
Conflict of interest
Xing Tian, Li-Ping Guo, Xiao-Long Hu, Jin Huang, Yan-Hua Fan, Tian-Shu Ren and Qing-Chun Zhao declare that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tian, X., Guo, LP., Hu, XL. et al. Protective Effects of Arctium lappa L. Roots Against Hydrogen Peroxide-Induced Cell Injury and Potential Mechanisms in SH-SY5Y Cells. Cell Mol Neurobiol 35, 335–344 (2015). https://doi.org/10.1007/s10571-014-0129-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-014-0129-7