
Abstract
Cellulose, which comprises D-glucose and L-glucose (D,L-cellulose), was synthesized from D-glucose (1D) and L-glucose (1L) via cationic ring-opening polymerization. Specifically, the ring-opening copolymerization of 3-O-benzyl-2,6-di-O-pivaloyl-β-D-glucopyranoside (2D) and 3-O-benzyl-2,6-di-O-pivaloyl-β-D-glucopyranoside (2L), synthesized from compounds 1D and 1L, respectively, in a 1:1 ratio, afforded 3-O-benzyl-2,6-di-O-β-D,L-glucopyranan (3DL) with a degree of polymerization (DPn) of 28.5 (Mw/Mn = 1.90) in quantitative yield. The deprotection of compound 3DL and subsequent acetylation proceeded smoothly to afford acetylated compound 4DL with a DPn of 18.6 (Mw/Mn = 2.08). The specific rotation of acetylated compound 4DL was + 0.01°, suggesting that acetylated compound 4DL was optically inactive cellulose triacetate. Furthermore, before acetylation, compound 4DL was an optically inactive cellulose comprising an almost racemic mixture of D-glucose and L-glucose. Compound 4DL was an amorphous polymer. This is the first reported synthesis of optically inactive D,L-cellulose.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Natural cellulose, which is the main constituent of plant cell wall, is a chiral homopolymer consisting of D-glucose (denoted as D-cellulose hereafter). This chirality is an important feature of high-value-added D-cellulose applications, such as chiral separation (Francotte 1994; Okamoto and Yashima 1998; D’Orazio et al. 2019; Chankvetadze 2020), chiral nematic D-cellulose liquid crystals (Nishio et al. 2016; Almedia et al. 2018), and D-cellulose-based organocatalysts for asymmetric synthesis (Ikai et al. 2011; Yasukawa et al. 2015; Ranaivoarimanana et al. 2019). Recently, an enantiomer of D-cellulose (denoted as L-cellulose hereafter) consisting of L-glucose was synthesized by our group using cationic ring-opening polymerization (Yagura et al. 2020). D-Cellulose and L-cellulose are crystalline and optically active polymers owing to their homochiral structures.
Poly (L-lactic acid) (PLLA) is a widely known alternative biodegradable chiral homopolymer (Rasal et al. 2010, Castro-Aguirre et al. 2016). PLLA has two stereoisomers, namely, poly (D-lactic acid) (PDLA) and poly (D,L-lactic acid) (PDLLA), the latter of which is a copolymer of L-lactic acid and D-lactic acid, which have also been well studied (Nampoothiri et al. 2010; Tsuji 2016). The structure and properties of PDLLA have been reported to be significantly different from those of PLLA and PDLA, as PDLLA is a diastereomer of PLLA or PDLA. For example, PDLLA with 15% L-lactic acid content is amorphous (Rasal et al. 2010). Therefore, cellulose consisting of D-glucose and L-glucose (denoted as D,L-cellulose hereafter) is expected to have a different structure and properties compared with D-cellulose. In particular, D,L-cellulose with a racemic mixture of D-glucose and L-glucose is expected to be an amorphous and optically inactive polymer owing to its racemic heterochiral structure. As the effect of D-cellulose chirality on its applications has not been well clarified, this optically inactive control sample is important for basic research on high-value-added applications of D-cellulose based on its chirality. However, to our knowledge, D,L-cellulose has not been found in nature or synthesized artificially. Our group has already reported the syntheses of D-cellulose (Nakatsubo et al. 1996; Kamitakahara et al. 1996; Adelwöhrer et al. 2009) and L-cellulose (Yagura et al. 2020) using cationic ring-opening polymerization. Therefore, this study describes the synthesis of D,L-cellulose with a racemic mixture of D-glucose and L-glucose (D-glucose/L-glucose = 1:1) as an optically inactive cellulose via a synthetic route using cationic ring-opening polymerization, as shown in Scheme 1.

Synthetic route for the preparation of D,L-cellulose (4DL) and D-cellulose (4D)
Experimental
Materials
D-glucose (1D) and L-glucose (1L) were purchased from Nacalai Tesque Inc. (Kyoto, Japan) and Tokyo Chemical Industry Co. (Tokyo, Japan), respectively, and were dried in a vacuum desiccator under high vacuum (less than 1 hPa) at 70 °C for 16 h before use. Compounds 2D and 2L were synthesized from compounds 1D and 1L, respectively, according to previous reports (Adelwöhrer et al. 2009; Yagura et al. 2020). Powdered 4 Å molecular sieves were activated in a GTO-2000 glass tube oven (Sibata Scientific Technology Ltd, Tokyo, Japan) under high vacuum (less than 1 hPa) at 300 °C for 3 h and kept at 105 °C in an SA46 drying oven (Masuda Co., Osaka, Japan) before use. All other chemicals were purchased from Nacalai Tesque Inc. and FUJIFILM Wako Pure Chemical Co. (Osaka, Japan), and used without further purification unless otherwise noted.
Measurements
Specific rotations were determined using a JASCO P-2200 polarimeter (JASCO, Hachioji, Japan) as the average values of five measurements. 1H and 13C NMR spectra were recorded on a Varian 500 NMR spectrometer (500 MHz, Agilent Technologies, Santa Clara, CA, USA) using tetramethylsilane (TMS) as an internal reference standard in CDCl3 as solvent, unless otherwise stated. Chemical shifts (δ) and coupling constants (J) are reported in ppm (parts per million) and Hz, respectively. Fourier-transform infrared (FT-IR) spectra were recorded in KBr pellets using a Shimadzu IRPrestige-21 spectrophotometer (Shimadzu, Kyoto, Japan). Gel permeation chromatography (GPC) was performed on a Shimadzu LC-10 system (Shimadzu) equipped with a Shimadzu UV–vis detector (SPD-10AVp) and a Shimadzu RI detector (RID-10A) under the following conditions: columns, Shodex K-802, K-802.5, and K-805 (Showa Denko K.K., Tokyo, Japan) arranged in series; eluent, CHCl3; temperature, 40 °C; flow rate, 1.0 mL/min; standards, polystyrene standards (Showa Denko K.K.). X-ray diffractograms were recorded on a Rigaku RINT-Ultima IV diffractometer (Rigaku, Tokyo, Japan). Samples were annealed at 120 °C for 1 h and then at 210 °C for 10 min under high vacuum (less than 1 hPa) before X-ray diffraction using a Sibata GTO-2000 glass tube oven.
3-O-Benzyl-2,6-di-O-pivaloyl-(1 → 4)-β-D,L-glucopyranan (3DL).
Compound 2D (100 mg, 0.24 mmol) and compound 2L (100 mg, 0.24 mmol) were placed in a glass ampoule in a vacuum desiccator under high vacuum (less than 1 hPa) at room temperature for 16 h. Anhydrous CH2Cl2 (200 μL; distilled from P2O5) and BF3−Et2O (2.9 μL, 5 mol%) were added via syringe through the sealed cap of the glass ampoule. The reaction mixture was stirred under atmospheric pressure at 37 °C for 24 h, after which solidification was observed. After the reaction, the mixture was dissolved in EtOAc (20 mL). The organic layer was washed with distilled water, saturated aqueous NaHCO3 solution, and brine, dried over anhydrous Na2SO4, and concentrated to afford compound 3DL as a colorless solid in quantitative yield.
Compound 3DL: \({\left[\alpha \right]}_{D}^{25} =\) -0.11° (c = 1.0 in CDCl3); DPn = 28.5 (Mw/Mn = 1.90); 1H NMR (CDCl3) δ 7.32–7.20 (CH2Ph), 4.95–3.17 (CH2Ph, H-1, H-2, H-3, H-4, H-5, H-6a, H-6b), 1.29–0.99 (C(CH3)3); 13C NMR (CDCl3) δ 178.0, 177.5, 177.3, 176.4 (C = O), 138.7, 137.3, 128.5, 128.4, 128.0, 127.8, 127.4, 127.3, 126.9, 126.8 (CH2Ph), 100.0, 99.3 (C-1), 83.2, 80.5 (C-3), 76.7, 76.0 (C-5), 74.5 (CH2Ph), 73.2, 73.1 (C-4), 72.8, 72.2 (C-2), 63.6, 63.0, 62.5, 61.5 (C-6), 38.8, 38.7 (C(CH3)3), 27.2, 27.1, 27.0, 27.0 (C(CH3)3).
(1 → 4)-β-D,L-Glucopyranan (D,L-cellulose, 4DL)
Compound 3DL (200 mg) was dissolved in THF/AcOH (5 mL; 1:1, v/v), and Pd(OH)2 on charcoal (100 mg) was added. The reaction mixture was hydrogenated with vigorous stirring under H2 at atmospheric pressure and room temperature for 24 h, filtered through Celite 535RVS (Nacalai Tesque Inc.), washing with THF, and concentrated azeotropically with EtOH to give the partially debenzylated product as a colorless solid. This product was subjected to further debenzylation under the same conditions three times to obtain the fully debenzylated product as a colorless solid. The product was dissolved in THF/MeOH (15 mL; 10:1, v/v) and 28% NaOMe in MeOH solution (0.2 mL) was added. The reaction mixture was stirred at 50 °C for 12 h and then neutralized with 1 M HCl aqueous solution. The resulting mixture was evaporated to remove THF and MeOH, dialyzed against distilled water using a Spectra/Por Dialysis Membrane (MWCO 100–500, Repligen Co., Waltham, MA, USA) for 4 days, changing the water every 12 h, and then freeze-dried to afford compound 4DL (54.6 mg, 72.7% yield) as a colorless solid.
Compound 4DL: FT-IR (KBr) ν 3404, 2903, 1634, 1558, 1373, 1362, 1154, 1069, 1026, 893 cm−1.
Compound 4DL was acetylated with Ac2O/pyridine at 50 °C for 12 h, and subjected to specific rotation, GPC and 1H/13C NMR measurements.
Acetylated compound 4DL: \({\left[\alpha \right]}_{D}^{25} =\) +0.01 o(c = 0.55 in CDCl3); DPn = 18.6 (Mw/Mn = 2.08), 1H NMR (CDCl3) δ 5.07 (H-3), 4.81, 4.76 (H-2), 4.56 (H-1), 4.42 (H-1, H-6a), 4.04, 4.02 (H-6b), 3.73, 3.72 (H-4), 3.50 (H-5), 2.14,2.10, 2.08, 2.01, 1.98, 1.95 (CH3); 13C NMR (CDCl3) δ 170.5, 170.4, 170.2, 169.8, 169.6, 169.3 (C=O), 100.5, 100.4 (C-1), 76.1, 74.8, 74.4, 73.7, 72.8, 72.3, 72.0, 71.9, 71.6, 71.3, 68.1, 67.7 (C-2, C-3, C-4, C-5), 62.8, 62.1, 61.5, 61.3 (C-6), 20.8 (× 2), 20.7, 20.6, 20.5, 20.5 (× 2) (CH3).
Results and discussion
Cationic ring-opening copolymerization of glucose orthoester derivatives 2D and 2L.
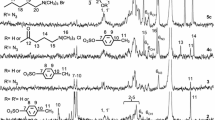
Glucose orthoester derivatives 2D and 2L were synthesized from compounds 1D and 1L, respectively, according to previous reports (Adelwöhrer et al. 2009; Yagura et al. 2020). The ring-opening copolymerization of compounds 2D and 2L in a 1:1 ratio was conducted to give compound 3DL in quantitative yield. Compound 3D was also prepared as a control by the ring-opening polymerization of compound 2D in a previous paper. Figure 1 shows the 1H and 13C NMR spectra of compounds 3DL and 3D. Signals assigned to benzyl groups in the range of 7.20–7.32 ppm and pivaloyl groups in the range of 0.99–1.29 ppm were found in the 1H NMR spectrum of compound 3DL, and signals assigned to benzyl groups in the range of 127–139 ppm and pivaloyl groups around 39 and 27 ppm were found in the 13C NMR spectrum of compound 3DL. Signals derived from acetyl groups in the range of 0.99–1.29 ppm and benzyl groups in the range of 127–139 ppm of compound 3DL were more complicated than the corresponded signals of compound 3D. It might be heterochiral structure of compound 3DL. Signals assigned to ring protons of anhydroglucose units (AGUs) and benzyl protons in the range of 3.17–4.95 ppm in the 1H spectrum of compound 3DL were more complicated than those of compound 3D. Similar results were observed for the 13C NMR spectra of compounds 3DL and 3D.

1H and 13C NMR spectra of compounds 3D and 3DL
Figure 2 shows HSQC NMR spectra of the AGU ring proton region in compounds 3DL and 3D. All correlation signals derived from compound 3D were observed, with new correlation signals observed in the NMR spectrum of compound 3DL. For example, the correlation signal derived from C-1/H-1 at 100.0/4.30 ppm, which corresponded to that from C-1/H-1 in compound 3D, and a new correlation signal at 99.3/4.72 ppm were observed. The chemical shifts of both signals suggested that only β-bonds were formed in the polymerization. There are theoretically four types of glycosidic linkage, namely, those between D-glucose and D-glucose (D–D), L-glucose and L-glucose (L–L), D-glucose and L-glucose (D–L), and L-glucose and D-glucose (L–D). The former two are homochiral structure linkages, while the latter two are heterochiral structure linkages. In the 1H and 13C NMR spectra, signals derived from C-1 and H-1 in pseudo-disaccharides with a D–D linkage have been reported to appear at different positions to those of C-1 and H-1 in pseudo-disaccharides with a D–L linkage (Ogawa et al. 1991; Duus et al. 1994). The correlation signals derived from C-1/H-1 in compound 3D (D–D linkages) and an enantiomer of compound 3D (L–L linkages) have previously been reported to appear at the same position (Yagura et al. 2020). In the NMR spectrum of compound 3DL, the correlation signal at 100.0/4.30 ppm was assigned to C-1/H-1 in the homochiral structure, while that at 99.3/4.72 ppm was assigned to C-1/H-1 in the heterochiral structure. The specific rotation of compound 3DL was − 0.11°. The results suggested that copolymerization proceeded smoothly to afford a β-glucopyranan derivative, with the composition ratio of compound 3DL almost reflecting the ratio of compounds 2D and 2L used. The DPn of compound 3DL was 28.5 (Mw/Mn = 1.90).

HSQC NMR spectra of compounds 3D and 3DL
Conversion of polymer 3DL into (1 → 4)-β -D,L-glucopyranan (4DL).
Compound 3DL was deprotected according to a previously reported method (Yagura et al. 2020), affording compound 4DL in 72.7% yield. Figure 3 shows the FT-IR spectra of compound 3DL, the debenzylated product, and compound 4DL. Bands attributed to benzyl groups at around 698 cm−1 and pivaloyl groups at around 1745 cm−1 had disappeared in the FT-IR spectrum of compound 4DL. Furthermore, compound 4DL was acetylated with Ac2O/pyridine at 50 °C for 12 h to give fully acetylated compound 4DL for characterization.

FT-IR spectra of compound 3DL (a); debenzylated 3DL (b); compound 4DL (c)
Figure 4 shows the 1H and 13C NMR spectra of acetylated compound 4DL. Signals assigned to benzyl groups, in the range of 7.20–7.32 ppm and around 128 ppm, and to pivaloyl groups, in the range of 0.99–1.29 ppm and around 39 ppm, also disappeared in the 1H and 13C NMR spectra of acetylated compound 4DL. These results suggested that the protecting groups in compound 3DL were completely removed. Signals derived from acetyl groups around 2.0 ppm were complicated because of heterochiral structure.

1H and 13C NMR spectra of acetylated compound 4DL
Figure 5 shows the HSQC NMR spectra of the AGU ring proton region in acetylated compounds 4D and 4DL. The signal pattern of acetylated compound 4DL was simplified compared with that of compound 3DL. This might be due to the bulky benzyl and pivaloyl groups being substituted with small acetyl groups. All correlation signals derived from acetylated compound 4D were observed, while new correlation signals were observed in the NMR spectrum of acetylated compound 4DL. In particular, correlation signals derived from C-1/H-1 in compound 4DL were observed at 100.5/4.42 ppm and 100.4/4.56 ppm. Correlation signals derived from C-1/H-1 in acetylated compound 4D were observed at 100.5/4.41 ppm in Fig. 5a and in commercial cellulose triacetate (Yagura et al. 2020). The H-1 signals in pseudo-cellobiose octaacetate with D–D 1,4-linkages and D–L 1,4-linkages have been reported to be observed at 4.52 and 4.67 ppm, respectively (Ogawa et al. 1991). The correlation signal at 100.5/4.42 ppm was assigned to C-1/H-1 in a homochiral structure, while that at 100.4/4.56 ppm was assigned to C-1/H-1 in a heterochiral structure in the NMR spectrum of acetylated compound 4DL. All correlation signals in the HSQC NMR spectrum of acetylated compound 4DL were assigned by the comparison with HSQC NMR spectra of acetylated compound 4D and acetylated compound 4DL with D/L ratio of 9/1 (Figure S1) which will be published in detail elsewhere. The specific rotation and DPn of acetylated compound 4DL were + 0.01° and 18.6 (Mw/Mn = 2.08), respectively. These results suggested that before acetylation, compound 4DL was a β-1,4-D,L-glucopyranan (D,L-cellulose) comprising an almost racemic mixture of D-glucose and L-glucose, namely, optically inactive D,L-cellulose.

HSQC NMR spectra of acetylated compounds 4D and 4DL
X-ray diffractograms of compounds 4DL and 4D are shown in Fig. 6. The diffractogram of compound 4DL indicated an amorphous structure like that of PDLLA, although it was annealed before measurement. This was due to the heterochiral structure of compound 4DL.

X-ray diffractograms of compounds 4D and 4DL
Conclusion
The ring-opening copolymerization of compounds 2D and 2L in a 1:1 ratio was conducted to give compound 3DL with a DPn of 28.5 (Mw/Mn = 1.90) in quantitative yield. Deprotection of compound 3DL and subsequent acetylation proceeded smoothly to afford acetylated compound 4DL with a DPn of 18.6 (Mw/Mn = 2.08). The specific rotation of acetylated compound 4DL was + 0.01°, suggesting that before acetylation, compound 4DL was an optically inactive cellulose. Compound 4DL was an amorphous polymer, as expected. This method for the synthesis of compound 4DL with a D/L ratio of 1:1 could be applied to the synthesis of D,L-celluloses with various D/L ratios. D,L-celluloses are expected to provide future research opportunities regarding the chirality of D-cellulose.
References
Almeida APC, Canejo JP, Fernandes SN, Echeverria C, Almeida PL, Godinho MH (2018) Cellulose-based biomimetics and their applications. Adv Mater 30:1703655. https://doi.org/10.1002/adma.201703655
Adelwöhrer C, Takano T, Nakatsubo F, Rosenau T (2009) Synthesis of 13C-perlabeled cellulose with more than 99% isotopic enrichment by a cationic ring-opening polymerization approach. Biomacromol 10:2817–2822. https://doi.org/10.1021/bm9006612
Chankvetadze B (2020) Recent trends in preparation, investigation and application of polysaccharide-based chiral stationary phases for separation of enantiomers in high-performance liquid chromatography. TrAC, Trends Anal Chem 122:115709. https://doi.org/10.1016/j.trac.2019.115709
D’Orazio G, Asensio-Ramos M, Fanali C (2019) Enantiomers separation by capillary electrochromatography using polysaccharide-based stationary phases. J Sep Sci 42:360–384. https://doi.org/10.1002/jssc.201800798
Duus JØ, Bock K, Ogawa S (1994) An NMR spectroscopic conformational study of 12 pseudo-disaccharides (D-glucopyranosyl-5a-carba-D- and -L-glucopyranoses. Carbohydr Res 252:1–18
Francotte E (1994) Contribution of preparative chromatographic resolution to the investigation of chiral phenomena. J Chromatogr A 666:565–601. https://doi.org/10.1016/0021-9673(94)80419-2
Ikai T, Moro M, Maeda K, Kanoh S (2011) Synthesis of polysaccharide derivatives bearing pyridine N-oxide groups and their use as asymmetric organocatalysts. React Funct Polym 71:1055–1058. https://doi.org/10.1016/j.reactfunctpolym.2011.07.010
Kamitakahara H, Hori M, Nakatsubo F (1996) Substituent effect on ring-opening polymerization of regioselectively acylated α-D-glucopyranose 1,2,4-orthopivalate derivatives. Macromolecules 29:6126–6131. https://doi.org/10.1021/ma960488h
Nakatsubo F, Kamitakahara H, Hori M (1996) Cationic ring-opening polymerization of 3,6-Di-O-benzyl-α-D-glucose 1,2,4-orthopivalate and the first chemical synthesis of cellulose. J Am Chem Soc 118:1677–1681. https://doi.org/10.1021/ja953286u
Nampoothiri KM, Nair NR, John RP (2010) An overview of the recent developments in polylactide (PLA) research. Biores Technol 101:8493–8501. https://doi.org/10.1016/j.biortech.2010.05.092
Nishio Y, Sato J, Sugimura K (2016) Liquid crystals of cellulosics: fascinating ordered structures for the design of functional material systems. Adv Polym Sci 271:241–286. https://doi.org/10.1007/12_2015_308
Ogawa S, Sugawa I, Shibata Y (1991) Synthesis of pseudo-laminarabiose, -cellobiose, and -maltose (D-glucopyranosyl 5a-carba-D- and L-glucopyranoses. Carbohydr Res 211:147–155. https://doi.org/10.1016/0008-6215(91)84153-6
Okamoto Y, Yashima E (1998) Polysaccharide derivatives for chromatographic separation of enantiomers. Angew Chem Int Ed 37:1020–1043. https://doi.org/10.1002/(SICI)1521-3773(19980504)37:8%3c3C1020::AID-ANIE1020%3e3E3.0.CO;2-5
Ranaivoarimanana NJ, Kanomata K, Kitaoka T (2019) Concreted catalysis by nanocellulose and proline in oragnocatalytic michael addittions. Mol 24:1231. https://doi.org/10.3390/molecules24071231
Rasal RM, Janorkar AV, Hirt DE (2010) Poly(lactic acid) modifications. Prog Polym Sci 35:338–356. https://doi.org/10.1016/j.progpolymsci.2009.12.003
Tsuji H (2016) Poly(Lactic acid) stereocomplexes: A decade of progress. Adv Drug Deliv Rev 107:97–135. https://doi.org/10.1016/j.addr.2016.04.017
Yagura T, Ikegami Y, Kamitakahara H, Takano T (2020) Synthesis of an enantiomer of cellulose via cationic ring-opening polymerization. Cellul 27:9755–9766. https://doi.org/10.1007/s10570-020-03512-z
Yasukawa T, Miyamura H, Kobayashi S (2015) Cellulose-supported chiral rhodium nanoparticles as sustainable heterogeneous catalysts for asymmetric carbon-carbon bon –forming reactions. Chem Sci 6:6224–6229. https://doi.org/10.1039/c5sc02510a
Acknowledgments
This work was supported by Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Challenging research 18K19230.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Human and Animal rights
This is not research involving Human participants and Animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ikegami, W., Kamitakahara, H., Teramoto, Y. et al. Synthesis of optically inactive cellulose via cationic ring-opening polymerization. Cellulose 28, 6125–6132 (2021). https://doi.org/10.1007/s10570-021-03970-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-021-03970-z